3. Fragenrunde Flashcards
Thema Pharmakokinetik, wie ist der Zusammenhang zwischen HWZ, Cl und Vd ?
Hwz: Zeitspanne, in der die Konzentration eines Arzneistoffes auf die Hälfte des Ausgangswertes abfällt. Sie ist wichtig zur Findung des Dosieintervalls.
Cl: fiktives Plasmavolumen, das pro Zeiteinheit von Stoff befreit wird. Sie gibt Auskunft über die glomeruläre Filtrationsrate.
Vd: pharmakokinetisch fiktives Volumen, in dem der Arzneistoff verteilt sein müsste, wenn er überall in gleicher Konz vorläge, wie im Plasma.
Aber wie ist der Zusammenhang, wie hängt was voneinander ab ?
Cl und Vd beeinflussen die HWZ.

Was ist nicht lineare Kinetik?
Zeigt keine lineare Korrelation Dosis und Plasmaspiegel. Wenn die Dosis verdoppelt wird haben wir keine direkte Verdopplung der Plasmaspiegel. Gründe hierfür sind: Löslichkeitsprobleme am Resorptionsort.
Enzyminduktion oder –inhibition.
Sättigbarer Metabolismus.
Welche Arzneimittel kennen sie, die keine lineare Kinetik haben?
Carbamazepin CYP3A4 Induktor (AUTOINDUKTION)
Phenytoin CYP3A4 Induktor
Was ist der Kumulationsfaktor ?
Kumulation ist die Anhäufung eines Arzneistoffes nach Mehrfachapplikation, wenn eine neue Dosis gegeben wird, ehe die vorherige vollens ausgeschieden ist.
Er dient zur Abschätzung des Dosisintervalls und ist der Kehrwert des Verlustfaktors.
Wenn der Verlust gering ist, ist die Kumulation groß. Die Konzentrationen Cmax und Cmin des Steady-State sind berechenbar. Er ist abhängig von Verlustfaktor, der die Menge an Stoff gibt, die über bestimmte Zeit eliminiert wird.
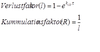
Welche Eigenschaften muß ein Arzneistoff haben um einer Elimination erster ordnung zu folgen?
Die verabreichte Dosis darf sich nicht linear zu den daraus folgenden Plasmaspiegeln verhalten. Dies ist der Fall z.B. beim sättigbaren Metabolismus ; enzyminhibition oder Induktion; Löslichkeitsprobleme am Resorptionsort.
Der Konzentrationsabfall pro Zeit ist nicht konstant, sondern verhält sich proportional zu der im Moment vorliegenden Plasmakonzentration, er ist also eine lineare Funktion der Plasmakonzentration. Dadurch dass pro Zeiteinheit ein konstanter Anteil der Plasmakonzentration ausgeschieden wird, nimmt die Plasmakonzentration zunächst schnell ab und mit sinkender Konzentration immer langsamer. Dies wird mit einer Exponentialfunktion beschrieben. Die meisten Pharmaka folgen einer Eliminationskinetik 1. Ordnung.
Erstellung eines Dosierschemas. Welche Parameter sind dafür wichtig?
Cmax, Cmin um den Dosisbereich und somit das therapeutische Fenster eingrenzen zu können. Das Dosierungsintervall ist abhängig von der HWZ, welche abhängig ist von der Eliminationskonstanten und dem Verteilungsvolumen. Die HWZ bestimmt direkt das Dosierungsintervall.

Wie quantitativ ist das abschätzbar, was wird durch das Dosierungsintervall verändert?
Die Steady-State Konzentration wird in ihrer Höhe verändert. Über die Veränderung des Dosierintervalls kann die Amplitude zw. Cmax und Cmin verändert werden. Folglich muß das Dosierungsintervall nach der Enge des therapeutischen Fensters gerichtet werden. Eine Gabe einer Initialen Maximalkonzentration gibt hierbei sehr schnell Plasmaspiegel, die dem des Steady State sehr ähnlich ist (ist noch kein Steady –State, da dieser per Definition nur über Mehrfachapplikation erreicht wird).
Die Erhaltungsdosis ist für die kontinuierliche Gabe wesentlich wichtiger, wobei diese durch die Clearance bestimmt wird.
Welche verschiedenen Möglichkeiten der Dosierungsstrategien gibt es?
Die Dosierung nach Körpergewicht, die eingeteilt wird in das tatsächliche und das ideale Körpergewicht. Das ideale Körpergewicht muß für Männer und Frauen nach einer unterschiedlichen Formel berechnet werden. Das größenbezogene Gewicht wird als ideal angesehen. Das Verteilungsvolumen eines Arzneistoffes mit geringer Bindung an endogene Strukturen bei Patienten mit normaler Konstitution erhöht sich mit zunehmendem tatsächlichem Körpergewicht (ist also abhängig von Hydrophilie und Lipophilie des Arzneistoffes). Gleichzeitig verschieben sich Wasser-, Muskel- und Fettgewichtsanteile. Es besteht kein linearer Zusammenhang zwichen dem tatsächlichen Gewicht und dem Verteilungsvolumen. Das ideale Körpergewicht würde die zusätzliche Verteilung ins Körperfett außer Acht lassen.
Wie verändert sich die Dosierung bei einer Nierenfunktionsstörung?
Es kann ein individueller Faktor errechnet werden. Dieser setzt sich zusammen aus dem Verhältnis K10 des nierenkranken/K10 des gesunden.
K10 des nierenkranken ist die Eliminationskonstante, die sich ergibt aus der Metabolisierung und der biliären Exkretion. Es ist somit der Anteil an nicht renal eliminierter Substanz. Aus dem Verhältnis ergibt sich dann der individuelle Korrekturfaktor Q .
Was ist das Verteilungsvolumen?
Das Verteilungsvolumen ist ein pharmakokinetischer Parameter, der darüber Aufschluss darüber gibt, wie stark sich ein pharmazeutischer Wirkstoff aus dem Blut in den extravasalen Raum und die Gewebe verteilt. Vd wird in L oder L/kg Körpergewicht angegeben.
Vd = D / C0
Das Verteilungsvolumen ist kein definierter anatomischer Raum im Organismus, sondern eine theoretische Grösse, welche eine Einschätzung darüber gibt, wie stark sich der Wirkstoff in die Gewebe verteilt. Grosse Verteilungsvolumina finden sich zum Beispiel bei lipophilen Wirkstoffen, da sich diese ins Fettgewebe verteilen. Umgekehrt haben polare Substanzen und Stoffe mit einer hohen Proteinbindung und einem hohem Molekulargewicht im Allgemeinen ein kleines Verteilungsvolumen, weil sie im Blut verbleiben. Übrigens: Ein grosses Verteilungsvolumen bedeutet nicht zwingend, dass sich der Wirkstoff auch zum Wirkort verteilt.
Vertieilungsvolumen in der terminalen Eliminationsphase
Verteilungsvolumen = Dosis/Plasmakonzentration
wenn Plasmakonzentration sinkt, steigt Verteilungsvolumen
Es werden dabei das zentrale Verteilungsvolumen Vc, in dem ein Medikament sich nach Aufnahme im Körper schnell verteilt, vom Verteilungsvolumen im steady state: Vss unterschieden, das nach der Verteilung im Körper eingenommen wird. Als Vz wird das Verteilungsvolumen in der terminalen Endphase der Elimination bezeichnet.
(Verteilungsvolumen steigt also bei geringerer Plasmakonzentration)
Clearance und deren Bestimmung
Die Clearance ist ein Maß für die Klär- bzw. Entgiftungsleistung der Nieren. Ihre Bestimmung dient der Überprüfung der Nierenfunktion. Sie entspricht dem rechnerischen Plasmavolumen pro Zeiteinheit, das von einem bestimmten Stoff geklärt wurde. Die Clearance wird in der Regel in ml/min angegeben.
Es gibt verschiedene Verfahren für die Messung der Clearance. Alle Solute, die zur Ermittlung der Clearance herangezogen werden, müssen die folgenden Eigenschaften aufweisen: Der Stoff darf im Tubulussystem und im Nierenmark
weder synthetisiert noch metabolisiert und
weder resorbiert noch sezerniert werden,
desweiteren
darf der Stoff die glomeruläre Filtrationsrate nicht beeinflussen und
muss im Glomerulus frei filtriert werden.
Dies trifft auf das im Folgenden genannte Kreatinin zwar nicht zu, weil es zu einer geringen Rate im proximalen Tubulus sezerniert wird. Dadurch liegt die Kreatininkonzentration um 10% höher als das filtrierte Solut. Da aber die Kreatininkonzentrationsmessung im Blut der gleichen Fehlerquote unterliegt, gleichen sich beide Fehler aus. Dennoch muß diese methodische Schwäche berücksichtigt werden, da bei unterschiedlichem Ausfallen der beiden Fehler ein tatsächlicher Fehler der Clearance-Berechnung resultieren kann.
Folgende Verfahren werden angewandt:
Die Inulin-Clearance misst das Filtrationsvermögen der Niere. Hierzu wird dem Patienten Inulin verabreicht und gemessen, wie viel vom verabreichten Stoff pro Zeit wieder ausgeschieden wird. Da Inulin zwar filtriert, nicht aber rückresorbiert wird, ist die Inulin-Clearance identisch mit der glomerulären Filtrationsrate (GFR). Für den gesunden Jugendlichen liegt der Wert bei etwa 125 ml/min. Eine Abnahme des Wertes deutet auf eine Störung in der Nierenfunktion hin. Mit zunehmenden Alter nimmt die GFR physiologisch auf 60-65 ml/min ab. Dies ist bei der Dosierung von Arzneistoffen, die über die Niere ausgeschieden werden, wichtig, da bei älteren Patienten durch die geringere GFR oft eine Verringerung der Dosis vorgenommen werden muss.
Die Kreatinin-Clearance wird wegen ihrer einfacheren Durchführung in der Klinik der Inulin-Clearance vorgezogen. Sie erlaubt bereits eine Bestimmung der GFR in guter Näherung. Es wird die Ausscheidung von Kreatinin gemessen, die annähernd der von Inulin entspricht. Die Kreatinin-Plasmaspiegel schwanken nur wenig, was diese Messung überhaupt erst möglich macht. Vorteilhaft ist weiterhin, dass die Infusion, die bei der Messung der Inulin-Clearance erforderlich ist, entfällt.
Die PAH-Clearance (Paraaminohippursäure-Clearance) dient der Bestimmung des renalen Plasmaflusses und des renalen Blutflusses.
Für spezielle Fragestellungen der Nierenfunktionsdiagnostik stehen in der Nuklearmedizin verschiedene Verfahren der Isotopen-Clearance zur Verfügung, die mit sehr hoher Genauigkeit
Halbwertszeit und terminale Halbwertszeit
Als Plasmahalbwertszeit, fallweise auch als Eliminationshalbwertszeit bezeichnet, definiert man diejenige Zeitspanne, die nach intravenöser Verabreichung zwischen der Maximalkonzentration eines Arzneistoffes im Blutplasma bis zum Abfall auf die Hälfte dieses Wertes verstreicht
Nach Applikation eines Stoffes laufen gleichzeitig Verteilungs- und Eliminationsprozesse ab, so dass man mehrere Plasmahalbwertszeiten ermitteln kann. In der Regel wird die längste (terminale) Halbwertszeit als Plasmahalbwertszeit angegeben.
nicht lineare Kinetik erklären
Eliminationskinetik 0. Ordnung
Syn. nicht-lineare Kinetik, dosisunabhängige Kinetik, kapazitätslimitierte Kinetik, nicht-übliche Kinetik
Pro Zeiteinheit wird eine konstante Menge eines Wirkstoffs eliminiert. Das bedeutet, dass der Konzentrationsabfall pro Zeit konstant und damit unabhängig von der im Moment vorhandenen Plasmakonzentration ist. Ein klassisches Beispiel ist die Elimination von Ethanol im menschlichen Körper.
Faktoren der nicht linearen Kinetik
Enzymausstattung und Zeit
Konzentration (konstant) / Zeit = Geschwindigkeitskonstante/Reaktionsgeschwindigkeit
Michaelis-Menten-Kinetik –> Parameter
v0 = vmax * [S] / Km + [S]
v0 gibt hierbei die initiale Reaktionsgeschwindigkeit bei einer bestimmten Substratkonzentration [S] an. vmax ist die maximale Reaktionsgeschwindigkeit.
Eine Kenngröße für eine enzymatische Reaktion ist die Michaeliskonstante Km. Sie hängt von der jeweiligen enzymatischen Reaktion ab. Km gibt die Substratkonzentration an, bei der die Umsatzgeschwindigkeit halbmaximal ist (v=½·vmax), die also bei Halbsättigung vorliegt
Dosierungsintervall
Wofür braucht man da die Halbwertszeit?
Abstand zwischen 2 Dosisverabreichungen
hängt ab von der Halbwertszeit, da ein

Erhaltungsdosis
Dosis zur Aufrechterhaltung bestimmter Plasmakonzentration bzw. bestimmten Plasmakonzentrationsbereichs
Ersatz der während des Dosierungsintervalls eliminierten Arzneistoffmenge
primär von Gesamtclearance abhängig
e.v.-Applikation sekundär von systemisch verfügbarer Fraktion
Überprüfung der Erhaltungsdosis auf Praktikabilität
Tabletten, Kapseln und Suppositorien nicht/nur bedingt teilbar
Patienten-Compliance berücksichtigen
ggf. praxisgerechte Modifizierung

Initialdosis
In akuten Fällen mögl. sofortiger Wirkeintritt
AS-Konzentration sofort im therapeutischen Bereich
dafür benötigte Arzneistoffmenge –> Initialdosis
Höhe abhängig vom scheinbaren Verteilungsvolumen

Empirische Dosisindividualisierung
Einfachste und im klinischen Alltag am häufigsten eingesetzte Methode
Empfehlungen, allgemeine und persönliche Erfahrungen des Arztes
Festlegung eines Dosierungsschemas, nach dem der Patient behandelt wird
Quelle: Arzneimittelfachinformationen, Dosierungstabellen
Größtenteils Ergebnisse klinischer Phase I-und –II-Studien der Arzneimittelentwicklung
Durchschnittswerte aus Untersuchungen an kleinerem, homogenem Kollektiv
Spätere Dosisänderungen
Pragmatisch: aufgrund des klinischen Bildes des Patienten
z.B. Dosiserhöhung/-reduktion bei nicht-eintretender erwünschter/Auftreten unerwünschter Wirkung
Individuell: mit Hilfe von Feedback-Kontrollen
Dosierung nach Körpergewicht
tatsächliches Körpergewicht (KG) oder Idealkörpergewicht (IKG)
IKG für Patienten, mit großem Einfluss des Gewichts auf Verteilung bzw. Elimination von Arzneistoffen
IKG = Körpergewicht, das bei Fettleibigen überschüssigen Fettanteil außer Acht lässt
Formeln nach Devine
Frauen IKG [kg] = 45,5 kg + 0,91 kg/cm ·(Körpergröße [cm] –152 cm)
Männer IKG [kg] = 50,0 kg + 0,91 kg/cm ·(Körpergröße [cm] –152 cm)
Bei Patienten normaler Konstitution steigt mit zunehmendem KG das Verteilungsvolumen (V) für lipophilen AS
Gleichzeitig verschieben sich Wasser-, Muskel- und Fettgewichtsanteile
Kein linearer Zusammenhang zwischen KG und V
Bei fettleibigen Patienten Änderung des auf das KG standardisierte Verteilungsvolumen (V in L/kg KG) möglich
Dosierung nach Körperoberfläche
Formeln zur Berechnung der Körperoberfläche nach Du Bois und Du Bois (1916)
KOF [m2] = KG [kg]0,425 · Körpergröße [cm]0,725 · 0,007184 [m2/kg/cm]
Für Kinder existieren andere Gleichungen!
Empirische Beobachtungen
Korrelation der KOF mit vielen physiologischen Parametern (Grundumsatz, Organgröße, Organleistung)
Bis heute für viele Arzneistoffe nicht belegt!
KOF-bezogene Dosierung von Zytostatika Standard
Einfache Bestimmungsmethoden im klinischen Alltag
Nur sehr grober Anhaltspunkt für individuelle Dosierungen
Andere patientenspezifische Faktoren müssen zusätzlich in Betracht gezogen werden
Was ist Q0? Wann ist wichtig, ob eine Substanz renal oder nicht-renal ausgeschieden wird?
Q0 = nicht renale Eliminationsgeschwindigkeitskonstante / Eliminationsgeschwindigkeitskonstante
wichtig bei Niereninsuffizienz

Was ist Q`?
Q` = (nicht-renale Eliminationskonstante + renale Eliminationskonstante) / Eliminationskonstante
individuelle Bestimmung von Dosis und/oder Dosierungsintervall mit Korrekturfaktor Q‘
Dosisreduktion bei gleich bleibendem Dosierungsintervall durch Multiplikation der Dosis eines Nierengesunden mit Q‘
Verlängerung des Dosisintervalls bei gleich bleibender Dosis durch Division von τ eines Nierengesunden durch Q‘
Im weiteren Therapieverlauf Plasmakonzentration überwachen und Dosis anpassen
Voraussetzungen zur Anwendung der Gleichungen und Graphiken zur Bestimmung von Q‘
Arzneistoffkonzentrations-Zeit-Verlauf : Einkompartiment-Modell
Elimination folgt Kinetik 1. Ordnung
Andere Prozesse wie systemische Verfügbarkeit, Proteinbindung und Verteilung bleiben unverändert

