Glycogen metabolism week 2 Flashcards
(26 cards)
What cellular compartment is glycogen stored in?
What tissues have the most glycogen?
- cytosol
- liver and skeletal muscle have the most glycogen.

What is the difference btwn the liver and skeletal muscle as it pertains to reasons for the storage of glycogen?
The glycogen in muscle is used as a fuel reserve for ATP synthesis while in the liver, it is used as a glucose reserve for the maintenance of blood glucose.

What do liver glycogen levels vary in response to?
How long do liver glycogen levels last during fasting?
Liver glycogen levels vary greatly in response to diet and feeding time. Glycogen accumulates to high levels after a meal and then slowly decreases between meals, supplying glucose to other tissues. Glycogen lasts 12-24 hours during fasting.

Why is most glucose from glycogen consumed within muscle cells without the formation of free glucose as intermediate?
What is the difference btwn the end products of glucose utilization in red and white muscle?
Which type of muscle has a greater capacity for glycogenolysis and glycolysis? What is the reason for this?
Muscle glycogen is used as a source of energy during increased muscle activity. Most of glucose from glycogen is consumed within muscle cells without formation of free glucose as an intermediate since muscle lacks glucose 6-phosphatase. About 8% is converted to free glucose within the tissue, but most of it is metabolized by glycolysis in muscle.
Thus, muscle glycogen does not help maintain blood glucose levels, as liver glycogen does.
Red muscle fibers are supplied with a blood flow, contain myoglobin and are packed with mitochondria. Glycogen mobilized within these cells is converted into pyruvate, which is oxidized to CO2 and H2O through the mitochondria.
In contrast, within white muscle fibers, the blood supply is poor and it has fewer mitochondria; glycogenolysis here supplies substrate for glycolysis with end product being lactate, thus producing less energy.
White muscle cells have enormous capacity for glycogenolysis and glycolysis, more than red muscle fibers.
White muscle cells can only function at full capacity for a relatively short period of time.

Glycogen consists mostly of glucosyl residues linked together by what 2 types of bonds?
How often do branches occur?
What end (reducing or non-reducing) are new residues linked to?
Why is glycogen branched?
Glycogen consists mostly of glucosyl residues linked together by alpha-1,4-glycosidic linkages. Branches occur through frequent alpha-1-,6-glycosidic linkages.
Branches occur at every fourth glucosyl residues within the central core and less frequently further out. Finally, glycogen is branched polysaccharide with several chains and several chain terminals. These “ends” will accept further glucose units during glycogenesis; or used as a site for stepwise degradation during glycogenolysis.
Why is glycogen branched? This gives more sites of degradation and synthesis so
the process can occur more rapidly.
Why is glycogen branched? This gives more sites of degradation and synthesis so the process can occur more rapidly.

What are 2 reasons for storing glucose as glycogen? (as opposed to storing it as fat or free glucose)
Why not fat? Glycogen can be mobilized faster in muscle than fat. Fat cannot be used as an energy source in the absence of oxygen and fat cannot be converted to glucose to maintain glucose plasma levels.
Why not glucose? We cannot store glucose because it is osmotically active and ATP would have to be used to pump glucose in the cell against the concentration gradient to reach about 400 mM equal to the glycogen content (0.01 microM). Storage as glycogen does not change the osmolarity as would free glucose.
What end of glycogen chains (reducing or non-reducing) does glycogenolysis happen?
What enzyme performs the first step of glycogenolysis (on amylose chains)? What cofactor is required? What is the difference btwn the way this enzyme works and how alpha amylase functions?
What is the second step of glycogenolysis?
The third step of glycogenolysis is tissue dependent. Explain the differences.
Step 1: Glycogen phosphorylase (B6 is its cofactor) catalyzes phosphorolysis of glycogen, a reaction in which Pi is used in the cleavage of an _alpha-1,4-glycosidic_ linkage between two glucose units to yield Glucose-1-P. Note that this does not require ATP (uses free inorganic phosphate). This always occurs at terminal non-reducing ends of glycogen chains. In contrast, alpha-amylase, a glycosidase present in the saliva and in the pancreatic juice, degrades glycogen and starch in the gut by simple hydrolysis using water rather than inorganic phosphate to cleave alpha-1,4-glycosidic bonds.
Step 2:
In the next step, phosphoglucomutase converts G-1-P to Glucose 6-P in a nearequilibrium reaction, which allows synthesis as well (reversible reaction).
Step 3:
The next step depends on the tissue:
In liver, glucose 6-phosphate produced by glycogenolysis is hydrolyzed by glucose 6 phosphatase to give free glucose.
In peripheral tissues, the G6P generated by glycogenolysis is used in glycolysis leading to the production of lactate in white muscle fibers and mostly to oxidation to CO2 in red muscle fibers.

What enzyme is needed to hydrolyze amylopectin branches of glycogen? Why is this enzyme needed? Explain how this enzyme works.
Step 4: A Debranching Enzyme is required for complete hydrolysis of glycogen
The first enzyme of glycogen degradation, glycogen phosphorylase is specific for alpha 1,4 glycosidic linkages. However, it stops at 4 glucose unit before the alpha 1,6-branch points resulting in what is called a phosphorylase limit dextrin. The debranching enzyme is a bifunctional enzyme that with its first activity (transferase), removes a strand of three glucosyl residues from a four glucosyl residue branch. The strand is then transferred to a free hydroxyl group of a glucosyl residue at the end of the same or adjacent glycogen molecule. This results in a longer amylose chain with only one glucosyl residue remaining in alpha-1,6-linkage (glycogen phosphorylase then resumes its activity on this now longer amylose chain). This remaining glucose in alpha-1,6 linkage is broken hydrolytically (by the second activity (glucosidase) and produces free glucose.

What is von Gierke’s disease type I? What are the 2 subtypes and what are they caused by?
What are the symptoms/consequences of this disease?
How is this disease treated?
The most common glycogen storage disease, referred to as type I or von Gierke’s disease, is caused by a deficiency of liver, intestinal mucosa, and kidney glucose 6- phosphatase. Thus diagnosis by small bowel biopsy is possible. Patients with this disease can be further subclassified into those lacking the glucose 6 phosphatase enzyme per se (type Ia) and those lacking the glucose 6-phosphate translocase (type Ib).
Clinical manifestations include fasting hypoglycemia, lactic acidemia, hyperlipidemia, and hyperuricemia with gouty arthritis. The fasting hypoglycemia is readily explained as a consequence of the glucose 6-phosphatase deficiency, the enzyme required to obtain glucose from liver glycogen and gluconeogenesis. The liver of these patients does release some glucose by the action of the glycogen debrancher enzyme. The lactic acidemia occurs because the liver cannot use lactate effectively for glucose synthesis (G6P is a gluconeogenic enzyme). In addition, the liver inappropriately produces lactic acid in response to glucagon. This hormone should trigger glucose release without lactate production; however, the opposite occurs because of the lack of glucose 6-phosphatase. Hyperuricemia results from increased purine degradation in the liver (consequence of higher synthesis); hyperlipidemia because of increased availability of lactic acid for lipogenesis and lipid mobilization from the adipose tissue caused by high glucagon levels in response to hypoglycemia. The manifestations of von Gierke’s disease can greatly be diminished by providing carbohydrate throughout the day to prevent hypoglycemia. During sleep this can be done by infusion of carbohydrate into the gut by a nasogastric tube.
see slide 20 of notes

What is Pompe’s disease (type II glycogen storage disease) caused by? What are the consequences of this disease?
Type II glycogen storage disease or Pompe’s disease is caused by the absence of α-1,4-glucosidase (or acid maltase), an enzyme normally found in lysosomes. The absence of this enzyme leads to the accumulation of glycogen in virtually every tissue. This is somewhat surprising, but lysosomes take up glycogen granules and become defective with respect to other functions if they lack the capacity to destroy the granules. Because other synthetic and degradative pathways of glycogen metabolism are intact, metabolic derangements such as those in von Gierke’s disease are not seen. The reason for extralysosomal glycogen accumulation is unknown. Massive cardiomegaly occurs and death results at an early age from heart failure.
What is Cori’s disease (type III glycogen storage disease) caused by? What are the consequences of this disease?
Also called type III glycogen storage disease, Cori’s disease is caused by a deficiency of the glycogen debrancher enzyme. Glycogen accumulates because only the outer branches can be removed from the molecule by phosphorylase. Hepatomegaly occurs, but diminishes with age. The clinical manifestations are similar to but much milder than those seen in von Gierke’s disease, because gluconeogenesis is unaffected, and hypoglycemia and its complications are less severe.
What is McArdle’s disease (type V glycogen storage disease) caused by? What are the consequences of this disease?
Also called the type V glycogen storage disease, McArdle’s disease is caused by an absence of muscle phosphorylase. Patients suffer from painful muscle cramps and are unable to perform strenuous exercise, presumably because muscle glycogen stores are not available to the exercising muscle. Thus the normal increase in plasma lactate (released from the muscle) following exercise is absent. The muscles are probably damaged because of inadequate energy supply and glycogen accumulation. Release of muscle enzymes creatine phosphokinase and aldolase and of myoglobin is common (myocyte death due to lack of energy); elevated levels of these substances in the blood suggest a muscle disorder.
What is Hers disease (type VI glycogen storage disease) caused by? What are the consequences of this disease?
Liver phosphorylase deficiency. Mild fasting hypoglycemia.
see pg 104 of course notes
What is required as a primer for glycogen synthesis? What is the function of this primer?
Glycogen itself is the usual primer since it is always present. The outer regions are removed and resynthesized more rapidly than the core. A polypeptide, glycogenin, functions as a primer. It is a self-glucosylating enzyme that uses UDP-glucose to link glucose to one of its own Tyr residues. The glycosylated glycogenin then serves as a primer.
Explain the 5 steps of glycogenesis.
Step 1:
The first reaction is glucokinase in hepatic tissue or hexokinase in peripheral tissue.
Glucose + ATP –> glucose 6-P + ADP
Step 2:
The second freely reversible reaction is catalyzed by phosphoglucomutase:
Glucose 6-phosphate –> glucose 1-phosphate
Step 3:
The next step is catalyzed by glucose 1-phosphate uridyltransferase (another name for this enzyme is UDP-glucose pyrophosphorylase) to activate glucose:
Glucose 1-P + UTP –> UDP-glucose + PPi
The reaction is driven by pyrophosphatase action on PPi + H2O –> 2 Pi . This is an example of linked reactions of linking an unfavorable with a favorable one.
Step 4: Glycogen Synthase
Glycogen synthase transfers activated glucosyl moiety of UDP-glucose to form a new glycosidic bond, α1-4 linkage, to the growing chain.
Step 5:
Once an amylose chain of at least 11 residues has been formed, a branching enzyme removes a block of about 7 residues from the growing chain and transfers them to another chain to make an alpha 1,6-linkage.
How many ATPs are required per glucose transfer to make glycogen?
What enzyme is required to reform UTP from UDP.
- Cost in ATP utilization: Each glucosyl residue addition requires 2 ATPs. The combination of glycogenolysis and glycolysis to lactate yields only 3 ATP per glucosyl residue. Thus, the combination of glycogen synthesis and glycogen degradation to lactate yields only one ATP. Although, glycogen storage is not very efficient but its function is to provide a fuel reserve that can be quickly used, regardless of the economy.
- UDP as a product is then converted back to UTP by nucleoside disphosphate kinase: UDP + ATP –> UTP + ADP
The regulation of what 2 glycogenic enzymes must be regulated opposite of one another?
In what 2 general ways are they regulated?
Both glycogen synthase and glycogen phosphorylase are subject to control by allosteric effectors and covalent modification. The latter is triggered by hormones.

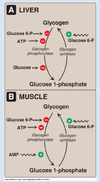
What are the allosteric regulators of glycogen phosphorylase activity? What are the differences btwn the regulation in liver and muscle?
liver: ATP, glucose, and glucose-6-phosphate inhibit glycogen phosphorylase.
muscle: ATP and glucose-6-phosphate inhibit glycogen phosphorylase. AMP and Ca2+ stimulate glycogen phosphorylase.
Note that free glucose is not an allosteric regulator of muscle glycogen phosphorylase. However, Ca2+ (which is necessary for musclular contraction) is a positive effector.
Also, note that ATP is not as strong of an effector for liver glycogen phosphorylase bc even if liver energy levels are low, glucose still may need to be released to the circulation.

What are the 2 forms of glycogen phosphorylase?
What 2 enzymes modify the activity of glycogen phosphorylase?
What are the effects of glucagon, epinephrine, and insulin on glycogen phosphorylase activity? How/why can we react to stress when starving or have low energy levels?
Can allosteric effectors act independently of covalent modification?
Phosphorylase is covalently modified by phosphorylation.
The phosphorylated (a) form is active and the nonphosphorylated (b) form is inactive.
The modifying enzymes are: Phosphorylase kinase and phosphoprotein phosphatase.
Phosphorylation causes a conformational change and turns the enzyme to phosphorylase a, which is active and unaffected by AMP.
Phosphorylase b (nonphosphorylated form) can be greatly activated by AMP. Thus, the covalent modification mechanism can be bypassed by the allosteric mechanism and vice versa.
Phosphorylase kinase itself is also subject to cyclic phosphorylationdephosphorylation through the glucagon/epinephrine –> cAMP –> PKA type mechanism. Protein kinase A (PKA) phosphorylates and activates phosphorylase kinase, which in turn phosphorylates and activates glycogen phosphorylase. Thus, stress or low glucose level triggers more phosphorylase a in order to obtain more energy. This is the reason that we can react to stress even when we are starving or low in energy levels.
When glucose supply is adequate, by the action of insulin, phosphoprotein phosphatase in turn dephosphorylates and inactivates phosphorylase kinase (and glycogen phosphorylase).

What is the allosteric effector of glycogen synthase? Does allosteric regulation of this enzyme vary btwn tissues?
Allosteric effectors: G-6P - excess glucose is available from glycolysis to store it away. This is the case in both liver and muscle.
What are the 2 forms of glycogen synthase?
How is glycogen synthase covalently modified?
What are the effects of glucagon, epinephrine, and insulin on glycogen synthase activity?
Glycogen synthase exists in a b form (inactive, phosphorylated), also called D for “dependent on the presence of G6P for activity”; and an a form (active, dephosphorylated form), also called I for independent of G6P. Note: In this case, phosphorylated is less active as opposed to glycogen phosphorylase.
Phosphorylation by kinases:
Phosphorylation of glycogen synthase through glucagon –> cAMP –> PKA inactivates the a form to b form. Thus, cAMP indirectly causes activation of phosphorylase and inactivation of synthase. Thus, stress will release glucose and shut off production of glycogen.
There are also cAMP independent, Ca2+ activated protein kinases and other kinases that are not subjected to regulation by cAMP or Ca2+. This allows control by more diverse signals. Calcium may also work through protein kinase C (PKC).
Dephosphorylation by phosphatases:
Just as with the phosphorylase phosphatase, there is a phosphoprotein phosphatase that converts phosphorylated glycogen synthase b back to glycogen synthase a. While cAMP promotes inactivation of synthase, insulin promotes activation.

In summary, how does glucagon effect glycogen degradation in the liver? How does it inhibit glycolysis in the liver?
What is the major fxn of glucagon in the liver?
Through what 2 receptors does epinephrine effect glycogen degradation in the liver? What are the cellular mechanims of these 2 receptors and how do they effect glycogen degradation?
Glucagon stimulates glycogen degradation and inhibits glycolysis at the levels of 6-phosphofructo-1 kinase and pyruvate kinase. This results in a very rapid increase in blood glucose. The major function of glucagon is to mobilize liver glycogen during periods of low food intake.
Epinephrine interacts directly with the Beta-adrenergic receptor in liver cells to activate adenylate cyclase to increase cAMP. It also inhibits glycolysis. Epinephrine can also interact with the α-adrenergic receptor to activate
phospholipase C to stimulates release of Ca2+ from the ER, which activates phosphorylase kinase, which activates glycogen phosphorylase and inactivates glycogen synthase.

What effect does epinephrine have on heart and skeletal muscle as it pertains to glycogen? Through what receptors does it influence glycogen stores?
What is the role of epinephrine in these tissues?
Epinephrine also stimulates glycogen degradation in the heart and in the skeletal muscle through β-adrenergic receptors that elevate cAMP. However, since heart and skeletal muscle lack glucose 6 phosphatase and since cAMP does not inhibit but rather stimulates glycolysis (remember PFK-2 regulation), the role of epinephrine in these tissues is to make more glucose 6-P available for stimulating glycolysis.

How does neural control effect glycogen degradation in skeletal muscle? (hint: involves calcium)
A nerve impulse causes membrane depolarization which in turn causes Ca2+ release from the sarcoplasmic reticulum into the sarcoplasma of muscle cells. This release of Ca2+ triggers muscle contraction while re-accumulation of Ca2+ by the sarcoplasmic reticulum causes relaxation. As Ca2+ levels increase there is more muscle activity and a greater need for ATP so the phosphorylase must be turned on. Activation of phosphorylase kinase by Ca2+ leads to activation of glycogen phosphorylase and, through additional kinases, the inactivation of glycogen synthase.



