Genetics 4 - Mitochondria Flashcards
learning outcomes

what is a mitochondrion
functions
genome
how did it originate
double membrane-bound organelle found in most eukaryotes
powerhouse of the cell - generate ATP
also involved in cell signalling
growth
death
cell cycle regulation
has independent (of nuclear DNA) circular genome
thought to have originated as free living bacteria which were taken up by eukaryotes to carry out ox phos (endosymbiotic theory)

no of mito. in nucleated cells
500-2000
cone cell photoreceptor of the eye - proportion of mito.
also muscles of eye
heart
mito. make up 80% of IC volume
60% - lateral rectus and other extra-ocular muscles
40% - heart
high proportion in energy dependent cells e.g. CNS, brain, eye, heart

human vs mitochondrial genome
- carriers of DNA
- sequences responsible for
- no of copies per cell
NUCLEAR GENOME
c. 23000 protein coding genes, 3200 Mb - million bps
23 de linear chromosomes (50-260 Mb each)
1.5% is protein coding sequences
non-functional gene related sequences (pseudogenes and gene fragments)
MITOCHONDRIAL GENOME
37 genes, 16.6 Mb
circular dsDNA (linear forms exist)
16569 bp (93% coding mRNA or tRNA or rRNA) - efficient
dozens of copies per mito. and 1000s of this genome/cell

mtDNA sequence
Cambridge sequence
no of genes of mito. genome
proportion that code for non mRNA
37 genes total
24 encode for non mRNA
22 mitochondrial tRNA (white)
1 mitochondrial 23S rRNA (blue)
1 mitochondrial 16S rRNA (blue)

no of genes transcribed and translated to proteins on mt ribosomes
what are they related to
13
all related to ox phos
7 NADH dehydrogenase subunits (ND - yellow) I
3 cytochrome C oxidase (COX - orange) IV
2 ATPase (ATP - red) V
1 cytochrome B (CYTB - peach) III

I
NADH dehydrogenase subunits
7 present
IV
cytochrome C oxidase
3 present
V
ATPase
2 present
III
cytochrome B
1 present
electron transport chain and ox phos
5 protein complexes
Pass protons along to create proton conc gradient
Protons pumped into space between inner and outer membranes
ATP produced

ROS generation and ATP synthase
ROS are generated during ATP synthesis
rate of O2 consumption has to correspond to rate of ATP synthesis so that ROS are neutralised
uncoupling of generation and consumption leads to accumulation of ROS - O2 ions and peroxides which are toxic
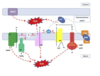
ROS uncoupling and accumulation
may occur in some mito. diseases especially if person has a fever
ROS accumulation ⇒ oxidative damage (including to mtDNA)
may trigger apoptosis
CNS may be particularly vulnerable to this
infection may trigger neurological problems in patients with mt disease

no of gene products needed to make a mitochondrion
where are mito. proteins encoded
3000 gene products to make a mito.
most mito. proteins are encoded on nuclear genome (transferred from mito. genome over evolutionary time) and synthesised in cytoplasm
therefore most inherited disorders of mito. are related to changes in nuclear DNA rather than mtDNA

no of mito. gene products needed to make ATP
13 - mtDNA
77 - nuclear genome
what remaining mito. gene products do
signalling molecules involved in regulation of:
MP
cell cycle control
development
apoptosis
cellular metabolism
how is mitochondrial DNA inherited
almost exclusively along maternal line
fertilised oocyte degrades mtDNA carried by sperm
mothers transmit mtDNA to sons and daughters
only daughters can transmit their mtDNA to the next generation
sons can inherit mtDNA disease but can not transmit

mtDNA sequence variation
mtDNA of any individual shows variation from Cambridge Consensus Sequence
most variation is silent polymorphisms

region on mtDNA - useful for forensic purposes
Control Region
highly polymorphic
mtDNA copies per cell
replication and cell cycle
100s-1000s of mtDNA copies per cell
variable - replication not coordinated with cell cycle
mtDNA vs genomic DNA - variation
reduced stringency of proofreading and replication error correction with mtDNA
no mtDNA repair mechanism
many fold higher sequence variation than genomic DNA
homoplasmy
all mtDNA sequences are the same



























