Chem II: 6-10 Flashcards
lewis acid
electron acceptor
tend to be electrophiles
vacant p orbitals that can accept electron pair
lewis base
electron donor
tend to be nucleophiles
have lone pair of electrons that can be donated
coordinate covalent bonds
covalent bonds in which both electrons in the bond came from the same starting atom (lewis base)
when lewis acid and bases interact
amphoteric
molecules that can act as either bronsted lowry acids or bases
ex: water
acid dissociatio constant
Ka
measures strength of an acid in solution
dissociation of acid HA
HA = H+ + A-
pKa of more acid molecules
smaller or negative pKa
pKa of more basic molecules
larger pKa
main functional groups that act as acids
alcohols, aldehydes and ketones (at alpha carbon), carboxylic acids and most derivatives
main functional groups that act as bases
amines and amides
nucleophiles
have either lone pairs or pi bonds that can form new bonds to electrophiles
tend to be good bases
strength based on rate of rxn - kinetic property
the more basic the nucleophile, the more ___ it is
reactive
factors that contribute to nucleophilicity
- charge: inc with inc electron density (more neg charge)
- electronegativity: dec as electroneg inc (bc less likely to share electron density)
- steric hindrance: dec as molecule is bulkier
- solvent: protoic solvents hinder
electrophile
- electron loving species with a positive charge or positively polarized atom
- accepts an electron pair when forming new bonds with a nucleophile
- usually act as lewis acids
- better leaving groups make it more likely that a rxn will happen
electrophilicity of carboxilic acid derivatives
most reactive to least: anhydrides, carboxilic acids, esters, amides
leaving groups
- retain the electrons after heterolysis
- stabilize extra electrons
- weak bases and conjugate bases of strong acids are GLGs
heterolytic rxns
- opposite of coordinate covalent bond formation
- a bond is broken and both electrons are given to one of the 2 products
nucleophilic subsitution reactions
SN1 and SN2
nucleophile forms a bond with a substrate carbon and LG leaves
SN1 steps
- LG leaves, making a carbocation: rate limiting step
- nucleophile attacks carbocation

SN1
- first order reaction
- loss of leaving group to make carbocation intermediate then nucl attack
- produce usually racemic mixture
- varied sterochemsitry bc nucl can attack C+ from either side
- most likely to occur on tertiary carbons (C+ easily stabilized)
SN2
- one step -> concerted rxn
- backside attack
- inversion
- stereospecific reaction
- 1° and 2° NOT 3° carbons
stereospecific reaction
configuration of reactant determines the configuration of the product due to the reaction mechanism
SN2 steps

how must the nucleophile and LG be related in order for a substitution reaction to proceed?
a substitution reaction will proceed when the nucleophile is a stronger base (more reactive) than the LG
what trends increase electrophilicity
greater positive charge snd better LGs –> make reaction more likely to proceed
features of GLGs
stabilize electra electrons
weak bases, resonance stabilization, electron withdrawing groups
chemoselectivity
preferential reaction of one function group in the presence of other functional groups
steric hindrance
prevention of reactions at a particular location within a molecule due to the size of substituent groups
SN1 vs SN2
mechanism
- SN2: single step, no intermediate, one transition state,
- lvg group departs as nucl adds (concerted)
- SN1: not single step, C+ intermediate (rate determining step)
- lvg grp leaves first –> C+ –> then nucl adds
SN1 vs SN2
kinetics
- SN2: bimolecular
- rate = k [nucl][RX]
- SN1: unimolecular
- rate = k [RX]
SN1 vs SN2
sterochemistry
- SN2: every substitution goes with inversion at carbon undergoing substitution
- SN1: mix of inversion and retention
- loss of configuration
SN1 vs SN2
electrophile scope
SN2:
- Best: Me-X
- Good: 1° R-X
- Possible: 2° R-X
- NO: 3° R-X
SN1:
- Best: 3° R-X
- Possible: 2° R-X
- Unlikely: 1° R-X
- NO: Me-X
SN1 vs SN2
solvent effect
- SN2: more polar solvent = rate inc or dec
- SN1: more polar solvent = inc rate
protection o fLGs
can temporarily maks a LG with sterically bulky group during synthesis
what are the 2 reactive centers of carbonyl containing compounds
carbonyl carbon -> electrophilic
alpha hydrogens -> acidic
what to consider for sterospecificity
consider whether configuration of reactant necessarily leads to specific configuration
what to consider for stereoselectivity
- if more than one product, major product will be determined by differences in strain or stability
- more striained molecules (with significant angle, torsional, or nonbonded strain) are less likely to form
- products with conjugation (alternating single and multiple bonds) are significant more stable
steps to problem solving
- nomenclature
- identify functional groups
- good nucleophiles, electrophiles, LGs, acid/bases
- identify other reagents
- same qs ^
- identify most reactive functional groups
- more oxidized arbons tend to be more reactive to both nucleo-electro rxns and redox rxns
- identify first step of rxn
- consider stereospecificity/stereoselectivity
alpha hydrogen
connected to alpha carbon
easily lost hydrogens –> acidic –> resonance stabilized conjugate base
alpha carbon
adjacent to carbonyl carbon
carbanion
molecule with negatively charged atom
alpha hydrogens of ketones vs aldehydes
alpha hydrogens slightly less acidic than aldehydes because of electron donating property of additional alkyl group in ketone –> destabilizes carbanion
aldehyde vs ketone reactivity
aldehydes slightly more reactive to nucleophiles than ketones because of steric hindrance in ketone –> makes higher energy, crowded intermediate
why are the alpha hydrogens of aldehydes and ketones acidic?
- inductive effects
- electronegative oxygen pulls electron density from C-H bond, weakening it
- resonance effects
- once deprotonated, resonance stabilization of neg charge between alpha carbon, carbonyl carbon, and electron withdrawing carbonyl oxygen increases stability
tautomers
constitutional isomers that differ from each other in the placement of a proton and the position of a double bond
enol
C-C double bond and alcohol
acidity at alpha carbon

enolate
conjugate base of enol
acidity at alpha carbon

enolization/tautomerization
process of interconverting from keto to enol tautomer

solutions to form enolate carbanion
common strong bases: OH, LDA, KH
michael addition
enolate attacks alpha beta unsaturated carbonyl
formation of new C-C bond
michael addition
electrophile
alpha beta unsat carbonyl
michael addition
nucleophile/michael acceptor
enol/enolate
michael addition
mechanism

kinetically controlled product
- formed more rapidly
- less stable
- favored with:
- rapid irreversible reactions
- lower temperatures
- strong, sterically hindered base
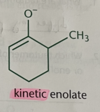
kinetic enolate
double bond on less substituted alpha carbon
thermodynamically controlled product
- formed more slowly
- more stable
- favored with:
- higher temperatures
- slow reversible reactions
- weaker, smaller bases
thermodynamic enolate
double bond with more substituted alpha carbon

enamination

imine
enamine
tautomers of imines

which tautomer of aldehydes and ketones is thermodynamically favored: keto or enol?
keto
aldol condensation
adding aldehyde/ketone to carbonyl of another
aldehyde or ketone acts as both electrophile and nucleophile
end result: C-C bond
aldol condensation
mechanism

aldol
molecule that condanes an aldhehyde and alcohol functional groups
condensation reaction
two molecules are joined with tthe loss of a small molecule
dehydration raction
water is lost
retro aldol reaction
aqueous base added and heat is applied
break bond between alpha and beta carbon of carbonyl
If you started with a chiral α-carbon, could tautomerization affect its chirality?
(A) Yes, because interconverting between the keto and enol forms can completely change the chirality to the opposite orientation (S->R).
(B) Yes, because interconverting between the keto and enol forms can create a racemic mixture of the α-carbon’s chirality center.
(C) No, because tautomerization is focused on the carbonyl carbon and cannot effect the α-carbon.
(D) No, because after any number of tautomerizations, the α-carbon will still retain its chirality.
(B) Yes, because interconverting between the keto and enol forms can create a racemic mixture of the α-carbon’s chirality center.
This effect is actually called α-racemization.
In an enolate formation reaction, the pKa of the α-hydrogen is 16.42 and the pKa of the resulting alcohol is 19.89. What is the Keq for this reaction? Will this reaction favor products or reactants?
(A) 29,512
(B) 2,951.2
(C) 295.12
(D) 29.512
(B) 2,951.2
pKeq = pKa(r) - pKa(p) pKeq = 16.42 - 19.89 pKeq = -3.47
Keq = 10^-pKeq Keq = 10^3.47 Keq = approx. 4700 (actual: 2,951.2) Thus, products are HIGHLY favored.
Why is the pKa of a ketone higher than an aldehyde?
The Methyl group donates electron density toward the carbonyl group, making it less willing to accept electrons from the α-hydrogen.
Draw the energy diagram a reaction as it goes to the thermodynamic product and as it goes to the kinetic product.

Why is the thermodynamic enolate more stable?
The thermodynamic enolate is more stable because it is more substituted.
The kinetic product is favored by a ________ base at _______ temperatures. Why is this the case?
(A) Bulky, High
(B) Bulky, Low
(C) Small, High
(D) Small, Low
(B) Bulky, Low
The kinetic product is favored by a bulky base at low temperatures. This is because the kinetic product is not sterically hindered, which results in a low activation energy; thus you only need a low temperature.
The thermodynamic product is favored by a ________ base at ________ temperatures. Why is this the case?
(A) Bulky, High
(B) Bulky, Low
(C) Small, High
(D) Small, Low
(C) Small, High
The thermodynamic product is favored by a small base at high temperatures. This is because the thermodynamic product is sterically hindered, which results in a high activation energy; thus you need a high temperature to overcome this.
Draw the Aldol Addition Reaction Mechanism in the presence of base.

Draw the Retro-Aldol Reaction Mechanism.


a


b


d


d


d


c


d


c


d

nomenclature
when aldehyde is attached to a ring
-carbaldehyde
dipole moment of carbonyl carbon of aldehydes and ketones
oxygen pulls electrons away from carbon –> makes carbon electrophilic and good target for nucleophiles
pyridinium chlorochromate (PCC)
used to make aldehydes thourgh partial oxidation of primary alcohol
name the compounds

butanone, propanal
given an alkane, an aldehyde, and an alcohol with equal length carbon chains, which will have the highest BP? why?
lowest to highest BP: alkane, aldehyde, alcohol
BP of aldehyde is elevated by its dipole, but the BP of alcohol is further elevated by hydrogen bonding
method for forming aldehyde
oxidation of primary alcohols
using weaker and anhydrous oxidizing agents like PCC (otherwise will fully oxidize to carboxylic acid)
method for forming ketones
oxidation of secondary alcohols
using reagents suchas sodium, potassium dichromate salts, CrO3, PCC
carbonyl carbon during reactions
electrophile -> ripe for nucleophilic attack
- when nucleophile attacks, forms covalent bond to carbon -> breaks pi bond in carbonyl
- electrons from pi bond pushed onto oxygen
- broken pi bond forms tetrahedral intermediate
- if no GLG
- carbonyl does not reform
- O- accepts proton from solvent to form hydroxyl group -> alcohol
- if GLG present
- carbonyl double bond can reform
nucleophilic addition reaction mechanism

hydration reaction
with aldehydes and ketone
mechanism
form geminal diols
nucleophilic oxygen in water attacks electrophilic carbonyl carbon

hemiacetal structure

acetal structure

hemiacetal formation
mechanism
one equiv of alc is added to aldehyde or ketone

acetal and ketal formation
mechanism
two equiv of alcohol added

imine formation
mechanism

HCN
hydrogen cyanide
classic nucleophile

cyanohydrin
structure

cyanohydrin formation
mechanism

when HCN reacts with an aldehyde or ketone, what functional group is produced? is the product stable?
cyanohydrin is produced, which is a stable product
Draw 4-bromopentanal.

Draw Cyclohexane Carbaldehyde

Draw 1,5-pentanedial

Draw (E)-4-hexen-2-one.

Draw 2-methylcyclohexanone.

Which of the following oxidizing agents would be able to convert an aldehyde to an alcohol?
(A) PCC
(B) PDC
(C) HIO4 (Periodic acid)
(D) None of the above
(D) None of the above
For the reaction in the question to occur, there must be a reducing agent! PCC, PDC and HIO4 are all weak oxidizing agents that can turn an alcohol into an aldehyde/ketone.
Order the following functional groups in order of increasing boiling point:
I. Isopropyl Alcohol
II. Acetone
III. Propane
IV. Propanal
(A) I < II < III < IV
(B) IV < III < II < I
(C) IV < II < III < I
(D) III < IV < II < I
(D) III < IV < II < I
In order of increasing boiling point: Propane (London Dispersion Forces only) < Propanal (Dipole-dipole Interactions) < Acetone (Dipole-dipole Interactions) < Isopropyl Alcohol (Hydrogen Bonding)
Why do Aldehydes have lower boiling points than Ketones?
The alkyl groups are electron-donating, which will make the carbons have a greater partial-positive charge and the oxgen have a greater partial-negative charge.
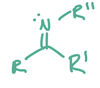
Functional groups with Nitrogen can be very good nucleophiles, and when added to carbonyls, can form either imines or enamines. Draw the general structure of both imines and enamines.

Draw the Hemiacetal Formation Reaction Mechanism in water.

Draw the Hemiacetal Formation Reaction Mechanisms in base.

Which of the following will shift the acetal mechanism to the right toward more product?
I. Add water
II. Increase amount of Aldehyde
III. Increase amount of Alcohol
(A) I Only
(B) I and II Only
(C) II and III Only
(D) I and III Only
(C) II and III Only
The following will shift the acetal mechanism to the right toward more product: REMOVING water (a product), increasing amount of aldehyde (a reactant), increasing amount of alcohol (also a reactant).
One final key nucleophilic additions to carbonyls is with Cyanide. Draw out the mechanism for creating a cyanohydrin with acid.


b


a


c


b


d


c


b

how oxidized are aldehydes in comparison to carboxylic acids and alcohols?
more oxidized than alcohols and less oxidized than carboxylic acids
oxidation of aldehydes with something stronger than PCC forms…
form carboxylic acids

oxidation reagents for making aldehydes to carboxylic acids
potassium permanganate (KMnO4), chromium trioxide (CrO3), silver (I) oxide (Ag2O), hydrogen peroxide (H2O2)
reduction of aldehydes and ketones forms…
alcohols

reduction reagents from making aldehydes and ketones into alcohol
hydride reagents
LiAlH4, NaBH4
what functional group is formed when an aldehyde is oxidized? what are some common oxidizing agents that assist this reaction?
functional group: carboxylic acid
common oxidizing agents: KMnO4, CrO3, Ag2O, H2O2
what functional group is formed when aldehydes and ketones are reduced? what are some common oxidizing agents that assist this reaction?
functional group: alcohol
common reducing agents: LiAlH4, NaBH4
A chemistry student reacts butanone with PCC and KMnO4. What are the expected products of each reaction?
Butanone reacts with neither because ketones cannot be oxidized with common oxidizing reagent.
A chemistry student reacts butanal with PCC and KMnO4. What are the expected products of each reaction?
Butanal is oxidized by KMnO4 to form butanoic acid, but does not react with PCC, which is not a strong enough oxidant.
Your lab instructor asks you to reduce the carboxylic acid of a compound that contains both a carboxylic acid and a ketone. You run the reaction, but end up with two alcohol groups instead of an alcohol group and a ketone. What important step did you forget?
You forgot to protect your ketone group by turning it into an ketal. A ketal would have been resistant to the reducing agent.
An Enamine is composed of what two functional groups?
(A) Amide, Alkane
(B) Amide, Alkene
(C) Amine, Alkane
(D) Amine, Alkene
(D) Amine, Alkene
An Enamine is composed of an amine and an alkene functional group.
Draw the reaction mechanism for Sodium Borohydride (NaBH4) reacting with an aldehyde.
Why would this reaction not work as well with NaH?
NOTE: A similar mechanism is seen with LiAlH4.
The previous reaction would not work well with H- because H- is a good base, but not a good Nucleophile.

True or false? The strong reducing agents often work by adding a proton (H+) to a carbonyl.
False. the strong reducing agents often work by adding a HYDRIDE (H-) to the carbonyl.
Why does Lithium Aluminum Hydride (LiAlH4) need to be used in dry conditions whereas Sodium Borohydride (NaBH4) does not?
LiAlH4 will react with water to form H2 gas, which could be dangerous. NaBH4 is not as reactive, so it isn’t an issue.
Why is Sodium Borohydride (NaBH4) less reactive than Lithium Aluminum Hydride (LiAlH4)?
Aluminum is less electronegative than Boron, resulting in greater electron density on the hydrogen for LiAlH4 (larger dipole moment).
LiAlH4 is strong enough to reduce all of the following functional groups, however, NaBH4 is only strong enough to reduce:
I. Aldehydes
II. Ketones
III. Carboxylic Acids
IV. Esters
(A) I Only
(B) II Only
(C) I and II Only
(D) I, II, III, and IV
(C) I and II Only
NaBH4 is strong enough to reduce Aldehydes and Ketones but not Carboxylic Acids and Esters.

a


c


d


d


d

phenol
aromatic ring
phenol nomenclature
two groups on adjacent carbons: ortho-, o-
two groups separated by a carbon: meta-, m-
two groups on opposite sides of the ring: para-, p-
alcohol physical properties
capable of intermolecular hydrogen bonding –> higher MP and BP, increased solubility in water
phenol physical properties
- aromatic ring –> resonance stabilized -> more acidic than other alcohols
- hydrogen bonding -> higher MP and BP
- slightly soluble in water
electron withdrawing groups ____ acidity
increase
electron donating groups ____ acidity
decrease
presence of more alkyl groups in nonaromatic alcohols produces ____ acidic molecules bc
less
alkyl groups donate electron density -> destabilize negative charge, stabilize positive charge
_____ alcohols can be oxidized to aldehydes using PCC
primary
jones oxidation
oxidizes primary alcohols to carboxyilic acids and secondary alcohols to ketons
reagent: CrO3, H2SO4

mesylate
compounds containing SO3CH3

tosylates
contain functional group -SO3C6H4CH3

mesylates and tosylates as protecting groups
protect when we do not want alcohols to react
will not react with other agents that would attack alcohols, especially oxidizing reagents
acetal
primary carbons with two -OR groups and a hydrogen atom
ketal
secondary carbons with two -OR groups
acetals and ketals as protecting groups
protect carbonyls from reacting with strong reducing agents like LiAlH4

what will happen to primary alcohols in the presence of strong oxidizing agents?
completely oxidized to carboxylic acids
what will happen to secondary alcohols in the presence of strong oxidizing agents?
can only be oxidized to ketones
what is the product when 1-butanol is treated with PCC?
aldehyde 1-butanal
what is the product when 1-butanol is treated with chromium trioxide?
carboxylic acid butanoic acid
how can aldehydes or ketones be protected using alcohols
can be reacted with 2 equiv of alc oh a diol to form an acetal or ketal
can be reverted back to carbonyl by catalytic acid
quinone
resonance stabilized electrophiles
used in ETC

treatment of phenols with oxidizing agents produces…
quinones

examples of biochemically relevant quinones
vitamin K1 (phylloquinone) and vitamin K2 (menaquinone
hydroxyquinone
ring with 2 carbonyls and variable number of hydroxyl groups
behave like quinones with EDGs
slightly less electrophilic than quinones (still quite reactive)
hydroquinone
benzene ring with 2 hydroxyl groups
ubiquinone
aka coenzyme Q
- vital electron carrie associated with Complexes I, II, and II of the ETC
- can be reduced to ubiquinol upon acceptance of electrons
- lipid soluble bc of long alkyl chain

how are quinones generally produced?
oxidation of phenols
how are hydroxyquinones produced?
oxidation of quinones
what chemical properties of ubiquinone allow it to carry out its biological functions?
- conjugated rings -> stabilize molecule when accepting electrons
- long alkyl chain -> lipid solubility -> allows molecule to function in phospholipid bilayer
Draw 1,4-Benzenediol (hydroquinone).

Draw 2-Bromophenol.

The Jones Reagent consists of Na2Cr2O7, H2SO4, and H2O. When these compounds are mixed they result in the formation of Chromic Acid. Draw the structure of Chromic Acid.

True or False? CrO3, H3O+, and Acetone may also be mixed to form Chromic Acid.
True. CrO3, H3O+, and Acetone may also be mixed to form Chromic Acid.
Draw the structure of Pyridinium Chlorochromate (PCC).

Draw the SN2 reaction of HBr reacting with an alcohol.

Draw the SN1 reaction of HBr reacting with an alcohol.

Draw the reduction of Benzoquinone to Hydroquinone.


b


b

