Exam 2: Platelets and Coagulation Flashcards
Platelet
Functions
- Limit bleeding after injury to a blood vessel
- Promote vessel repair
- Regular maintenance of an intact endothelium
Platelet Count
Normal range: 150,000 - 450,000 platelets/microliter
Thrombocytopenia
-
Abnormally low platelet count
- < 150,000/microliter
-
Characterized by:
- Easy bruising
- Nose bleeds
- Petechial rash
- Spontaneous bleeding
- Occurs below 20,000 platelets/microliter
Idiopathic Thrombocytopenic Purpura
(ITP)
Autoimmune disease where anti-platelet Ab destroy platelets.
Platelet
Lifecycle
- Derived from bone marrow cells called megakaryocytes
- Lifespan ~ 8-10 days
- Removed by sleen or via clotting process

Platelet
Morphology
Small, flat, disc-shaped membrane-enclosed bits of cytoplasm.
Hyalomere
- Clear outer region
- Contains bundles of microtubules
- Helps maintain discoid shape
- Contains actin & myosin
- Involved in shape change of activated platelets
Granulomere
- Central region with basophilic stippling
- Contains usual cytoplasmic organelles
- Contains at least 3 types of granules
- Alpha, Delta, and Lambda
2 systems of membrane bound channels:
-
Open canalicular system
- Invaginations of the plasma membrane
- Facilitates rapid exocytosis of granules
-
Dense tubular system
- Stores Ca2+ needed for exocytosis
- Not continuous with the plasma membrane

Alpha (α) Granules
Contains:
Platelet-derived growth factor (PDGF) ⇒ mitogen for vessel repair
von Willebrand Factor (vWF) ⇒ mediates platelet adhesion to endothelium (collagen and laminin)

Delta (δ) Granules
Contains:
Ca2+, ATP, ADP ⇒ all enhance platelet aggregation
Serotonin ⇒ vasoconstriction
(Picked up by platelets in circulation)

Lambda (λ) Granules
Contains:
Lysosomal enzymes ⇒ clot resorption

Vessel Repair
Process
Adhesion
-
vWF binds to components of the damaged basement membrane (collagen, laminin)
- vWF can be secreted by many cells including platelets
- vWF attracts platelets which have surface receptor for vWF
- Single layer of platelets forms @ site of endothelial damage
Aggregation
- Adhered platelets secrete fibrinogen
- Other platelets attracted to the site via cell surface receptors for fibrinogen
- Fibrinogen links the platelets together
-
Forms a multilayered primary hemostatic plug
- Fills the defect in the vessel wall
Activation
- Aggregation causes platelet activation
- Results in:
- Secretion of granule mediators
-
Synthesis and release of aracidonic acid derivatives
- Thromboxane A2 (TXA2)
-
Change of platelet shape
- Mediated by the hyalomere
- Released mediators cause:
-
Further platelet aggregation
- TXA2, serotonin, and Ca2+
-
Vasoconstriction (limits bleeding)
- Serotonin and TXA2
-
Blood coagulation
- Platelets release several coagulating factors from α-granules
- Meshwork of fibrin formed which stabilizes the platelet plug forming the secondary hemostatic plug
-
Further platelet aggregation
Clot Retraction
- After ~ 1 hr, platelets contract due to actin-myosin interaction
- Platelet plug ↓ in size & flattens against vessel wall
- Helps to re-establish smooth blood flow
Clot Resorption
- Mediated partially by lysosomal enzymes from λ-granules
Vessel Repair
- Mediated by platelet-derived growth factor (PDGF) from α-granules
- PDGF is strongly mitogenic for cells needed to rebuild the vessel wall including:
- Endothelial cells
- Fibroblasts
- Smooth muscle

Platelet Activation
Regulation
-
Platelets activated upon binding to collagen and laminin of damaged basement membrane
- Not exposed in healthy vessels
- Healthy endothelial cells produce factors that inhibit platelet aggregation
- Ex. Prostacyclin I2 (PGI2) from arachidonic acid

Aspirin
Inhibits cyclooxygenase activity
- ↓ platelet function
- Prolongs bleeding times

Thrombosis
Formation of a clot (thrombus)
- Can cause serious damage
- Can lead to death if vessel occluded

Thrombus
Charactertistics
- Meshwork of fibrin + plug of activated platelets
- Clotting occurs in association with membranes
- Pathology:
- In coronary arteries ⇒ MI
- In cerebral arteries ⇒ ischemic stroke
- In peripheral arteries ⇒ claudication (leg pain with exercise) or amputation
- In deep veins ⇒ DVT
Embolus
A mass traveling through the circulation.
Can be a thrombus broken off a site of coagulation.
If thrombus lodges in the lung ⇒ pulmonary embolism (PE)
Blood Clotting
Overview
Initiated on the membranes of endothelial cells and platelets:
- Formation of a fibrin clot
- Formation of a platelet plug
- Vasoconstriction (eicosanoids, PGs, Txs)
- Limits to the process (anticoagulation)
- Clot dissolution (fibrinolysis)
- Wound repair
Functions through cascade of proteolytic cleavage or conformational changes.
Fibrin Clot Formation
Charactertistics
- Intrinsic and extrinsic pathways converge on the final common pathway
-
Major factors
- Named by Roman numerals & common names
- Factor IX = Christmas factor
- Are glycoproteins synthesized primarily by the liver
- Named by Roman numerals & common names
-
Functions using cascades
- Activation primarily by proteolytic cleavage
- Successive proteins are serine proteases
- Cleaves peptide bond on carboxyl side of Arg or Lys
- Activation can also be caused by conformational changes
- Facilitates acceleration and amplification of process
- Non-proteolytic proteins also needed ⇒ accessory proteins (cofactors)

Clotting Effectors
Presence accelerates the rate of certain steps in fibrin clot formation:
-
Negatively charged phospholipids (PS, PI)
- Normally found on inner leaflet of plasma membrane
- Exposure signals injury
-
Ca2+
- Binds negatively charged γ-carboxyglutamate (Gla) residues on certain clotting proteins
- Facilitates binding of these proteins to exposed negatively charged phospholipids

Gla Proteins
Factors II, VII, IX, X
- Contains negatively charged γ-carboxyglutamate (Gla) residues
-
γ-carboxylation
-
Post-translational modification
- 9-12 Glu residues @ N-terminus carboxylated to Gla residues by glutamyl carboxylase
- Occurs in lumen of RER in hepatocytes
-
Vit K required as co-enzyme
- Oxidized in reaction to epoxide
- Must be reduced back to hydroquinone form to continue
- Done by Vitamin K epoxide reductase (VKOR)
-
Post-translational modification

Dicumarol
&
Warfarin (Coumadin)
⊗ reduction of Vit K by VKOR
- Inhibits clotting by depleting pool of Vit K
- Oral administration
- Slow onset
- Long half-life
- Polymorphisms in cytochrom P450 and VKOR isozymes results in varied dosing

Tissue Factor
(TF)
-
Transmembrane glycoprotein
- Abundant in vascular subendothelium
- Released by damaged tissue ⇒ extravascular
-
Key protein in the extrinsic pathway
- Pathway is quickly shut down by tissue factor pathways inhibitors (TFPI)

Extrinsic Pathway
“Tissue Factor Pathway”
- Vascular injury exposes extravascular TF (Factor III) to Factor VII
- TF binding causes conformation change of VII activating it to VIIa
- VII can also be activated by Factor XIIa from intrinsic path or thrombin (IIa) from common path
- VIIa is a serine protease which activates factor X
- Factor Xa enters the common pathway
Extrinsic pathway is quickly shut down by tissue factor pathway inhibitors (TFPI).

Intrinsic Pathway
“Contact Pathway”
All protein factors are found in the blood ⇒ intravascular
Contact Phase
Results in the activation of factor XII → XIIa
-
Contact of blood with a negatively charged surface ⇒ conformational change & activation of factor XII → XIIa
- In vitro ⇒ glass blood vials
- Sodium citrate/oxalate added to chelate Ca2+ and prevent clotting
- In vivo ⇒ ⊖ PL on damaged endothelium or an abnormal surface
- E.g. mechanical heart valve, stent, knee/hip replacements
- In vitro ⇒ glass blood vials
-
Amplification of contact phase
-
Factor XIIa cleaves Prekallikrein-HMWK at an anionic surface producing Kallikrein
- HMWK = High molecular weight kininogen
- Kallikrein can then proteolytically cleave additional Factor XII ⇒ amplification
-
Factor XIIa cleaves Prekallikrein-HMWK at an anionic surface producing Kallikrein
No known bleeding disorders associated with factor deficiencies of the contact phase.
X Activation Phase
-
Factor XIIa cleaves XI-HMWK at an anionic surface to produce Factor XIa
- Factor XI can also be activated by Thrombin from common pathway
- Defective Factor XI → Hemophilia C
-
Factor XIa cleaves Factor IX (Christmas factor) → IXa
- Defective Factor IX → Hemophilia B
- Can also be cleaved by Factor VIIa from extrinsic pathway
-
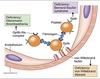
Factor IXa combines with Factor VIIIa
- Interaction with VIIIa ↑↑↑ rxn rate
- Factor VIII found in the blood bound to von Willebrand factor (vWF)
- vWF protects VIII from degradation
- Thrombin from common pathway cleaves vWF off VIII activating it
- Defective Factor VIII → Hemophilia A
-
IXa:VIIIa complex cleaves Factor X → Xa
- Factors VIIIa, IXa, 10, and Ca2+ on membrane ⇒ Tenase complex

Hemophilia
-
Coagulopathy caused by clotting factor deficiencies
- Factor VIII ⇒ Hemophilia A
- 6x more common than B
- Found on chromosome Xq
- Factor IX ⇒ Hemophilia B
- Found on chromosome Xq
- Factor XI ⇒ Hemophilia C
- Autosomal recessive
- Factor VIII ⇒ Hemophilia A
- Manifestations
- Decreased/delayed ability to clot
- Formation of abnormally friable clots
- Severity related to residual activity
- Effects seen if < 30% activity
- Severe form if < 1% activity
- Spontaneous, prolonged bleeding particularly into joints and muscle
- Treatment
- Recombinant factor replacement
- Somatic gene therapy in development