Blood and BM Path Chapter 18 - Acute Myeloid Leukemia Flashcards
(52 cards)
Adults tend to get ___ leukemias.
Kids tend to get ___ leukemias.
Adults tend to get myeloid leukemias.
Kids tend to get lymphoid leukemias.
WHO categories of AML
- AML with recurrent cytogenetic abnormalities
- AML with myelodysplasia-related changes
- AML with therapy-related changes (history of methylating agent or topoisomerase II inhibitor chemotherapy exposure)
- AML not otherwise specified
Why was the required BM blast percentage decreased from 30% to 20% in the 2001 edition of the WHO guidelines?
Because it has been demonstrated that “MDS-EBII” patients with greater than 20% blasts had a prognosis identical to those with AML.
Why is diagnosing APML such an emergency?
Because if it is not differentiated from another form of AML and the patient starts conventional chemotherapy, there is a high risk of DIC.
The only definitive morphologic finding that differentiates myeloid blasts from lymphoid blasts
Auer rods
Immature vs mature myeloid surface antigens
Immature: CD13, CD33, CD117
Mature: CD15
Megakaryocytic surface markers
CD41a and CD61
These are also surface markers for platelets!
“AML without maturation” vs “AML with minimal differentiation”
“without maturation”: <3% MPO, SBB-negative blasts, no Auer rods (which contain MPO and SBB!)
“with minimal differentiation”: >3% MPO, SBB-positive, Auer rods present
Subcategories of “acute myeloid leukemia with recurrent genetic abnormalities”
- Balanced translocations
- Inversions
- Mutations of NPM1 or CEPBA genes
Prognosis of various AML with recurrent genetic abnormalities

3 AML diagnoses that do not require 20% blasts in the marrow
- t(15:17) PML-RARa
- inv(16) OR t(16:16) CBFB-MYH11
- t(8:21) RUNX1-RUNX1T1
Recurrent genetic abnormalities that strattle the line between MDS and AML
inv(3), t(3:3), or t(6:9)
These are all associated with multilineage dysplasia and may be a cause of primary MDS, may present as MDS-EBSI or -EBSII, or may present as frank AML (>20% blasts) with multilineage dysplasia

- AML with RUNX1-RUNX1T1, Associated with t(8:21)
- RUNX1 encodes the core binding factor alpha protein (CBFA), one component of a heterodimeric hematopoietic TF. Abnormal fusion to RUNX1T1 (aka ETO for eight twenty one) impairs or misdirects gene regulatory activity.
- Morphologically appear with blasts displaying ample cytoplasm and abundant granules, sometimes with Auer rods. “Sunset cell” morphology is characteristic.
- Dyspoietic morphology of granulocytes is common (Pelger-Huetoid)
- Tell-tale features:
- Corkscrew Auer rods
- Prominent perinuclear hofs (green arrows)
- Large salmon pink granules (black arrow) intermixed with typical dark granules
- Together, the above make a “sunset cell”
- Expression profile:
- Frequent aberrant expression of B cell markers (CD19, Pax5, cytoplasmic CD79a)
- CD34 overexpression
- Myeloid profile: CD13, CD33, MPO
- Prognostics:
- High presenting WBC count (>20) - poor
- KIT mutation - intermediate


- AML with CBFB-MYH11, associated with inv(16) or t(16:16)
- Involve the gene for the second half of the core binding factor TF, CBFB. It is fused to a smooth muscle myosin heavy chain (MYH11).
- Tend to occur in younger patients
- Most (but not all) show granulocytic or monocytic blasts accompanied by abnormal eosinophil precursors within the marrow
- These eosinophil precursors show occasional to numerous large purple or basophilic granules of variable shape intermixed with eosinophilic granules
- Expression profile:
- Often abnormally express CD2
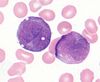
- Acute promyelocytic leukemia with PML-RARa. Associated with t(15:17)
- Known for life-threatening bleeding or clotting events in association with DIC
- If the diagnosis is even suspected, all trans retinoic acid is initiated even before confirmation
- Most common in middle-age. 2/3 of cases present with low WBC counts.
- Morpholoci features:
- “Hypergranular” morphology
- Abnormal promyelocytes with bilobed (“sliding discs”) nuclei
- Frequent Auer rods, often multiple per cell
- Expression characteristics:
- Promyelocytes often with CD34-HLA-DR- immunophenotype
- Bright CD33, MPO, and SBB
- Variable CD13
- May abberantly express CD64 or CD2
Less common genetic abberations of the retinoic acid receptor
-
ATRA-resistant
- t(11:17) ZBTB16-RARa
- t(17:17) STAT5B-RARa
-
ATRA-susceptible
- t(11:17) NUMA1-RARa
- t(5:17) NPM1-RARa
These are reported as “AML with a variant RARa translocation.” They also may have very different morphologies.

- AML with MLLT3-MLL, associated with t(9:11)
- Recently separated from the group of 11q23 MLL gene translocations to reflect its better prognosis (others have poor prognosis)
- MLL has histone methyltransferase activity. Likely it plays a role in gene regulation via epigenetic mechanisms.
- Usually seen in pediatric patients
- Morphologic features
- Blasts with monocytic or myelomonocytic morphology
- Somewhat bland, blasty appearance
- Surface expression
- No special patterns
- CD34- and CD117-
- MPO positive

- AML with DEK-NUP214, associated with t(6:9)
- DEK is a chromatin-associated protein and NUP214 a nuclear pore protein. The mechanism of the fusion’s contribution to oncogenesis has not been well described, but is thought to involve abnormal nuclear transport.
- Uncommon translocation that may affect a wide age range of patients. Prognosis is poor.
- Usually in the context of pancytopenia with myelodysplasia in all lineages
- Morphologic features:
- Distinguished by presence of BM or blood basophilia (>2% basophils), seen in 50% of cases
- Blasts may be myeloid or monocytic
- Surface expression:
- Unremarkable myeloid or monocytic immunophenotype

- AML with RPN1-EVI1, associated with inv(3) or t(3:3)
- EVI1 (ectopic virus integration-1) is a proto-oncogene with zing finger protein homology, proposed to have transcriptional repression activity via recruitment of histone deacetylases. Fusion to RPN1, a proteasome component, may serve to drive EVI1 overexpression.
- Relatively rare form of AML. Primarily occurs in adults. Prognosis is poor.
- Associated with myelodysplastic findings in granulocytes and platelets, usually with relative sparing of erythroids
- Patients may present before the blast count exceeds 20%
- Morphologic features:
- Presence of monolobated or bilobated megakaryocytes, which may be increased in number
- Blasts are myeloid, monocytic, or megakaryocytic
- Surface expression:
- Unremarkable myeloid blast immunopthenotype
- Megakaryoblastic cases express CD41 and CD61

- AML with RBM15-MKL1, associated with t(1:22). Often called “megakaryoblastic AML”
- RBM15 encodes a protein with RNA-binding motifs and MKL1 encodes a transcriptional coactivator of serum response factor (SRF). The mechanism of this translocation’s oncogenicity is unknown.
- Very rare form of infant leukemia. Presents as HSM or other organomegaly.
- Morphologic features:
- Megakaryocytic differentiation with medium-sized or larger cells that ahve round nuclei and fine reticular chromatin, visble nuclei, agranular basophilic cytoplasm, and occasional cytoplasmic blebbing (arrows)
- Marrow fibrosis is common (as with other megakaryoblastic leukemias)
- Surface expression:
- Blasts lacking CD45, CD34, and HLA-DR
- Express myeloid markers CD13 and CD33
- Express megakaryocyte markers CD41, CD61, and sometimes CD42
RTK mutations in AML
KIT and FLT3 are commonly mutated in AML, particularly in cases with normal karyotypes.
These take the form of internal tandem duplications and point mutations of the tyrosine kinase domain. They correlate with a poor prognosis.
KIT mutations are especially common in t(8:21) RUNX1-RUNX1T1 and inv(16), which herald a poor prognosis.

- AML with mutated NPM1 (with or without associated FLT3 mutation)
- NPM1 encodes a nuclear shuttling protein reported to have roles in robosome and centrosome biology as well as regulation of the ARF-TP53 pathway.
- More common in adult AML (30% of adult AML, 5% pediatric AML)
- Typically have a normal karyotype.
- Morphologic features:
- Myelomonocytic or monocytic
- No myelodysplastic findings
- May take the form of AML without maturation or AML with maturation and erythroleukopenia
- Blasts with cup-shaped nuclear invaginations are associated with this mutation, especially when it co-occurs with FLT3 mutations.
- Surface expression:
- Blast immunophenotype with loss of CD34
- Staining for NPM1 reveals overexpression on IHC
- Diagnosis
- Typically detected by PCR-based methods
- Exception is tetranuceotide insertion in exon 12 creating frameshift, which interferes with nuclear localization signaling and interrupts export
AML with no chromosomal anomalies, CD34+MHCII+, and aberrant expression of CD7. Erythropoiesis is preserved but patient has severe thrombocytopenia with purpura.
- AML with mutated CEBPA
- CEBPA is a TSG and transcription factor implicated in the differentiation of many disparate cell lineages, including granulocytes
- Detected in 10% of all AML cases, ~25% of cases co-occur with FLT3-ITD mutations
- No apparent association with age or sex.
- Relative preservation of erythropoiesis, but worse thrombocytopenia and higher circulating blast count. Extramedullary disease is less frequent.
- Morphologic features:
- FAB M1 (no maturation) and M2 (with maturation) blast morphology
- Rarer cases have myelomonocytic or monocytic morphology
- Surface expression:
- CD34+HLA-DR+
- Often aberrantly express CD7
- Diagnosis:
- Requires gene sequencing, as over 100 distinct mutations have been described

- AML with myelodysplasia-related changes
- Characterized by: Myeloid blast count >20% in the blood OR bone marrow of a patient WITHOUT history of cytotoxic chemotherapy/radiation therapy and WITH at least one of the following:
- Prior Hx of MDS or MDS/MPN
- Characteristic myelodysplasia-associated cytogenetic abnormalities
- Multilineage dysplasia in at least 50% of cells in at least 2 lineages
- Must not be explained by a recurrent genetic abnormality (this takes diagnostic precedence)
- Clinical features:
- Dominated by severe cytopenias
- Morphologic features:
- Neutrophils with hypogranularity, hyposegmented nuclei, or bizarre nuclear segmentation patterns
- Erythroid precursors with megaloblastoid changes (delayed nuclear maturation relative to cytoplasmic hemoglobinization), multiple nuclei, or irregular nuclear contours, ringed sideroblasts, cytoplasmic inclusions, PAS+ vacuoles
- Megakaryocytes with micromegakaryocyte morphology, hyposegmented nuclei, or widely separated nuclear lobes
- Blast morphology is variable, but AML with maturation and acute myelomonocytic leukemia morphologies are most common
- Surface markers:
- Limited utility
- Non-specific aberrancies such as expression of CD7 or TdT