Heritable Bleeding Disorders Flashcards
Describe the formation of a haemostatic plug
- Normal platelets in flowing blood
- Damaged endothelium leads to platelets adhering to the wall (bind to collagen in sub endothelium) and becoming activated
- Activated platelets aggregate to form a thrombus (platelet plug)
- Receptors are expressed and fibrinogen binds to platelets
- Granules are released, stimulating further platelet activation
- Coagulation cascade follows – clotting factors are soluble proteins and contribute to fibrin formation.

Which enzyme converts fibrinogen to fibrin?
thrombin (serine protease)
How is thrombin formed?
Prothrombin (coagulation factor II) is cleaved to form thrombin
What is Von Willebrand factor?
Von Willebrand factor helps platelets aggregate and adhere to (collagen in) walls of blood vessels.

What is the most common heritable bleeding disorder?
Von Willebrand disease
What is Von Willebrand disease?
low or dysfunctional Von Willebrand factor –> bleeding
What acts as receptors on platelet surfaces?
Membrane glycoproteins
The membrane glycoproteins of platelets act as receptors that mediate 2 important functions. What are these?
- adhesion to the subendothelial matrix
- platelet aggregation
What are the 2 important membrane glycoproteins found on platelets? What is each one a receptor for?
- 2b3a –> receptor for fibrinogen
- 1b9 –> receptor for vWf
Inside the platelets are granules. What are the 3 different types of granules found?
- alpha granules
- dense granules
- lysosomal granules
What do alpha granules contain?
contain adhesion proteins and calcium (role in coagulation cascade)
What do dense granules contain?
contain ATP and ADP which are released when platelets become activated (ADP forms positive feedback loops – activates more platelets)
Metabolic processes are going on inside cytoplasm of platelets when they become activated. What 2 major metabolic processes are going on?
- Formation of prostaglandins
- Formation of thromboxane
How are prostaglandins formed in the cytoplasm of platelets?
- Mobilisation of fatty acids from platelet membrane
- Conversion of the fatty acid arachidonic acid to intermediate prostaglandins
How is thromboxane formed in the cytoplasm of platelets?
- Synthesised by activated platelets from arachidonic acid (via the enzyme cyclooxygenase)
- Released from phospholipid membrane upon platelet activation
- Secreted into surrounding and stimulates activation of new platelets as well as platelet aggregation
What enzyme converts arachidonic acid to thromboxane?
cyclooxygenase
how does aspirin act as an anti-thrombotic agent?
Aspirin inhibits cyclooxygenase (COX) –> prevents production of thromboxane A2
Name 4 anti-platelet drugs
- Aspirin
- Clopidogrek
- Dipyridamole
- IIb and IIa anatagonists

How does Clopidogrel act as an anti-platelet drug?
- blocks ADP receptor (P2Y12 inhibitor)
- P2Y12 is a chemoreceptor for ADP
How does Dipyridamole act as an anti-platelet drug?
Stimulated prostacyclin to inhibit binding site for collagen, thrombin, thromboxane A2
How do IIb and IIa antagonist act as an anti-platelet drugs?
blocks GPIIb/IIIa
Overview of coagulation cascade
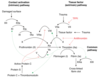
Describe the steps of the extrinsic pathway
- Coagulation is triggered by tissue factor
- Factor VII binds to tissue factor and becomes activated –> VIIa
- VIIa can directly activated X –> Xa

What is tissue factor? Where is it found?
a cellular protein found on surface of cells – damaged endothelium, inflamed blood vessels, WBCs, brain cells
Describe the steps of the intrinsic pathway
Doesn’t rely on molecules external from the blood but instead becomes activated when blood is exposed to a foreign surface:
- Factor 12 is activated when exposed to a foreign surface –> XIIa
- XIIa then activates XI –> XIa
- XIa then activates IX –> IXa
- IXa (with cofactor VIII) activates X –> Xa

Describe the steps of the common pathway of the coagulation cascade
- Xa (with cofactor Va) will convert prothrombin –> thrombin
- Thrombin converts fibrinogen to fibrin (crosslinked by Factor 13)

What is the ‘prothrombin time’?
- Used to measure extrinsic pathway:
- Adding a tissue factor substitute (thromboplastin) to plasma in a test tube
- Time how long it takes blood to clot
What is the Activated Partial Thromboplastin Time (APTT) used to measure?
Intrinsic pathway

What is the thrombin clotting time?
- Used to measure final step
- Thrombin is added to plasma
- Time how long it takes to clot
What is the thrombin clotting time dependent on?
- Very dependent on level of plasma fibrinogen
- Dependent on there being nothing in blood inhibiting formation of fibrin from fibrinogen
- E.g. heparin
What is often the first line of investigation of someone with abnormal bleeding?
Prothrombin Time (PT)
What does a coagulation screen comprise?
- Prothrombin time
- Activated Partial Thromboplastin Time (APTT)
- Thrombin Clotting Time
Why are the top parts of the intrinsic pathway is less physiologically important?
- Extrinsic pathway is thought to start it off.
- Intrinsic pathway acts as amplification system instead of initiator.
- Thrombin has positive feedback effect
Factor 11 can therefore be activated through extrinsic pathway activating thrombin and then the thrombin activating Factor 11 through a feedback system.

Does a patient with Factor VII deficiency have a bleeding disorder? What will the APTT be like?
No - this factor is less important
But will have a long APTT (instrinsic pathway time)
Will a patient with a factor XI deficiency have a bleeding disorder?
Yes but only mild
Protein C and Protein S also play a role in the coagulation cascade. What is their function?
Both have a role in anticoagulation:
- Protein C plays an important role in regulating anticoagulation by inhibiting Factor V
- Protein S has a role in the anticoagulation pathway, where it functions as a cofactor to Protein C in the inactivation of Factors Va and VIIIa
Antithrombin also has a role in the coagulation cascade. What is its function?
Anticoagulant effects; It inactivates several enzymes of the coagulation cascade, in particular thrombin and factor Xa
The fibrinolytic system also has a role in the coagulation cascade. What is its function?
Anticoagulant: The fibrinolytic system comprises an inactive proenzyme, plasminogen, which can be converted to the active enzyme, plasmin, which in turn degrades fibrin into soluble fibrin degradation products.
When conducting an assessment of a suspected bleeding disorder, clinical history is key.
What questions should you ask?
- Date of onset
- Previous history (longer is congenital, shorter if acquired)
- Clinical pattern (more severe presents in childhood)
- Response to challenges: surgery, dental extraction
- Young children: bleeding from umbilical stump, vaccinations, circumcision
- Requirement for medical/surgical intervention to ascertain extent of bleeding
- Systemic illness, drug history, family history – helps to identify if congenital
When conducting a clinical examination during an assessment of a suspected bleeding disorder, waht should you look for?
- Pattern of any bruising
- Signs of underlying disease: Joints, muscles, skin
Exampls of blood tests done during an assessment of a suspected bleeding disorder
- FBC and blood film
- Coagulation screen
- Clauss fibrinogen (D-dimer if suspicion of acquired disorder)
- Mixing studies: if clotting tests are longer than expected, they can be performed with the patient’s plasma and normal plasma. Test will correct significantly if there is a lack of factors. It will not correct significantly if the patient’s blood contains an inhibitor of clotting.
- Von Willebrand profile
- Coagulation factor assays
- Inhibitor assays (antiphospholipid type)
- Platelet function tests
- If all normal and suspicion remains: factor 13 and alpha2antiplasmin assays.
During mixing studies, if clotting tests are longer than expected, they can be performed with the patient’s plasma and normal plasma.
- If the clotting test corrects significantly, what does this indicate?
- If the clotting test does not correct significantly, what does this indicate?
- Lack of factors in patients blood
- Patients blood contains an inhibitor of clotting
If a patient has a normal coagulation screen (APTT, PT, TCT), what are other potential causes of a bleeding disorder?
- Thrombocytopenia (low platelets)
- Disorder of platelet function
- vWD
- Factor 13 deficiency
- Mild coagulation factor deficiency
- Vascular disorder
- Disorder of fibrinolysis (rare)
- Platelet/vessel wall defects (1ary haemostasis) –> give rise to prolonged bleeding time
How can a factor 13 deficiency lead to bleeding? How does this bleeding present?
- Factor 13 covalently crosslinks the fibrin polymer and stabilises it
- Patients with this deficiency typically present with late bleeding (a few days after sometimes)
How can platelet/vessel wall defects give rise to bleeding disorders?
- Reduced number of platelets (thrombocytopenia)
- Abnormal platelet function (could be iatrogenic, caused by aspirin)
- Abnormal vessel wall
- E.g. Ehlers-Danlos syndromes; a group of rare inherited conditions that affect connective tissue –> bleeding due to defects in collagen
- Abnormal interaction between platelets and vessel wall
- E.g. vWD – reduction in platelet adhering molecules to the wall.
What is Ehlers-Danlos syndromes?
Ehlers-Danlos syndromes; a group of rare inherited conditions that affect connective tissue –> bleeding due to defects in collagen
Difference in presentation between vascular/platelet defects and coagulation defects

What is this? What would it indicate?

Petechiae
- Little red spots often around hair follicles
- Don’t blanch with pressure
- Not palpable (flush against skin)
- Think about; thrombocytopenia, meningitis, vascular/platelet defects
There are 3 types of Von Willebrand Disease. How does each type differ?
- Type 1:
- Reduced production of normal vWf, tend to have normal/slightly reduced Factor VIII
- Common, mild; especially in blood group O
- Variable penetrance in a family with all the same genetic defect.
- Type 2:
- Patient is producing enough vWf but often is structurally abnormal so is dysfunctional (low molecular weight polymers)
- Slightly more severe than type 1
- Type 3:
- vWf is absent altogether and Factor VIII tends to be greatly reduced too
-
Severe and spontaneous;
- bleeding into joints and vessels as well as mucosal surfaces

Why can vWd often present with reduced Factor VIII levels?
Vwf is a carrier protein for factor VIII (carries it around circulation)
Inheritance pattern of vWf (remember different types)?
- Mainly autosomal dominant inheritance.
- Type 3 is autosomal recessive.
Which blood group is often associated with low vWf levels?
Blood group O (this is not vWd)
Symptoms of vWd?
- Postoperative/post-partum bleeding
- Mucocutaneous bleeding
- Menorrhagia
vWf has 2 functions. What are these?
- binds to protein 1b9 on the platelet surface and helps it bind to the vessel wall (collagen)
- forms a complex with factor 8 (carrier protein) to activate factor 10
What factors are deficient in vWd?
In vWD, there is less 8a, F10a and fibrin and so bleeding is more likely
Treatment options for vWd:
- Anti-fibrinolytics –> tranexamic acid
- DDAVP
- Factor concentrates containing vWF (pool plasma derived)
- Vaccination again hepatitis
- Combined OCP for menorrhagia (or Mirena coil)
What is tranexamic acid? What is it useful in treating?
Anti-fibrinolytic - good for bleeding of the mouth, periods and nosebleeds.
What is DDAVP? What is it used to treat?
- DDAVP is an analogue of vasopressin
- Analogue of vasopressin
- Side effect of promoting secretion of vWf from where it is stored inside endothelial cells inside lining of blood vessels –> short term solution
What is Factor I?
Fibrinogen
What is Factor II?
Prothrombin
Haemophilia A is a defect in what?
Factor VIII
Haemophilia B is a defect in what? What is this also known as?
Factor IX - Christmas disease
Summary of Heritable Coagulation Factor Deficiencies

Inheritance of haemophilias?
X-linked recessive

How does haemophilia affect men vs women? Why?
- Typically expressed in males and carried by females.
- In females, the normal X chromosome will compensate for the faulty X chromosome so tend to be carriers only
Is haemophilia A or B more common?
Haemophilia A: 1 in 5000 males
Haemophilia B: 1 in 30,000 males
Which part of the coagulation cascade is affected in haemophilia A/B?
8 and 9 are intrinsic pathway factors and contribute to the common clotting cascade.
Factor VIII or IX levels determine if haemophilia is mild, moderate or severe. What are the Factor VIII or IX levels for each of these?
- Normal: Factor VIII or IX level between 50-150%
- Mild: Factor VIII or IX level between 6-50%
- Moderate: Factor VIII or IX level between 1-5%
- Severe: Factor VIII or IX level less than 1%
How does mild haemophilia present?
- Bleeding only with moderate trauma or surgery.
- Diagnosis often due to family history or later in childhood
How does moderate haemophilia present?
Variable, occasional spontaneous bleeds, often proceeding trauma
How does severe haemophilia present?
Frequent bleeds into muscle and joints
Types of bleeding - reflect the severity of haemophilia
- Spontaneous/post traumatic
- Can be diagnosed after surgery, trauma, dental procedure etc
- Joint bleeding = haemarthrosis, sets of synovitis and causes abnormal, damaged joints
- Muscle haemorrhage – hallmark of severe haemophilia, leading to contractures, compartment syndrome, shortening, MSK disability
- Soft tissue – bleeds into tongue, pharynx, primary teeth eruption, can threaten the airways (life threatening).
- Life threatening bleeding – intracranial bleeding in severe/moderate
Treatment of haemophilia;
- Replacement of missing clotting protein (daily): on demand or prophylaxis.
- Using factor concentrates; recombinant are products of choice, no longer blood derived
- DDAVP (only for mild/moderate haemophilia A)
- Antifibrinolytic agents – for mucosal bleeding, GI bleeding
- Vaccination against hepatitis A and B
- Supportive measures: icing, immobilisation, rest
What are the supportive measures for haemophilia?
icing, immobilisation, rest
Which haemophilia does DDAVP treat?
A
One complication of treatment of haemophilia is the potential development of inhibitors. What are these?
Antibodies formed against Factor VIII or IX that stop the factor concentrates from working due to immune response:
- More common in haemophilia A (25%)
- Genetic predisposition
Presentation of development of inhibitors during haemophilia treatment?
- Poor recovery, bleeding difficult to manage, muscle/joint damage.
- Poor clinical response to treatment, factor ineffective or less effective
- Occurring early in course of condition, around 10 exposure days
Emicizumab is a treatment used for Haemophilia. Mechanism?
Used for haemophilia A only, with and without inhibitors
- Is a “monoclonal antibody”
- Monoclonal antibodies are proteins that are designed to bind to a specific target in the body
- They can be used to treat diseases or conditions
- Emicizumab:
- Mimics the action of the missing FVIII in the clotting pathway
- The cue for its action is FIXa, which appears after an injury
- Emicizumab binds to FIXa and FX, forming a link between these two clotting factors
- FIXa can then activate FX, allowing the pathway to continue and the blood to clot
Factor VIII Prophylaxis can also be used in haemophilia A treatment. How does this work?
- FVIII concentrate replaces missing or low levels of FVIII
- Prophylaxis is the regular administration of FVIII concentrate, to try and prevent bleeds
- FVIII rises quickly after infusion and then decreases over a number of hours
