2: Hematology Flashcards
Which 6 coagulation factors are vitamin K dependent?
- Factor II
- Factor VII
- Factor IX
- Factor X
- Protein C
- Protein S
[UpToDate: Depending upon the cause of deficiency, vitamin K can be administered in doses of one to 25 mg via oral, intramuscular, subcutaneous, or intravenous routes. When vitamin K deficiency occurs in patients who are also receiving coumarin-like anticoagulants, doses of vitamin K should be minimized in order to prevent refractoriness to further anticoagulation.
Vitamin K status can be determined indirectly by measuring vitamin K-dependent factors (ie, prothrombin, factors VII, IX, X, or protein C). In patients who are vitamin K deficient, levels of these factors often are less than 50% of normal. Measurement of des-gamma-carboxyprothrombin (DCP) in plasma is another more sensitive way of determining vitamin K deficiency. In normal subjects, DCP is zero; it is elevated in vitamin K deficiency from whatever cause and/or liver disease.]
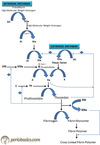
What are the 5 components of the prothrombin complex?
- Factor Xa
- Factor Va
- Calcium
- Platelet factor 3
- Prothrombin
[Medscape: Prothrombin complex concentrate (PCC) is an inactivated concentrate of factors II, IX, and X, with variable amounts of factor VII. Guidelines recommend the use of PCC in the setting of life-threatening bleeds, but little is known on the most effective dosing strategies and how the presenting international normalized ratio affects response to therapy.]
[Wikipedia: Prothrombin complex concentrate (PCC), also known as factor IX complex, is a medication made up of blood clotting factors II, IX, and X. Some versions also contain factor VII. It is used to treat and prevent bleeding in hemophilia B if pure factor IX is not avaliable. It may also be used in those with not enough of these factors due to other reasons such as warfarin therapy. It is given by slow injection into a vein.]

What are the 3 thrombolytic agents?
- Streptokinase (High antigenicity)
- Urokinase
- Tissue plasminogen activator (tPA)
[UpToDate: Recombinant tissue type plasminogen activator (tPA, alteplase), streptokinase (SK), and recombinant human urokinase (UK) are the best studied thrombolytic agents for the treatment of acute PE, that are approved by the US Food and Drug Administration (FDA). Other thrombolytic agents include lanoteplase, tenecteplase, and reteplase. The characteristics of SK, tPA, and UK are described briefly here, with greater detail presented elsewhere.
tPA is a naturally occurring enzyme produced by a number of tissues including endothelial cells. tPA binds to fibrin, which increases its affinity for plasminogen and enhances plasminogen activation.
SK is a polypeptide derived from beta-hemolytic streptococcus cultures. It binds to plasminogen, forming an active enzyme that activates plasmin. Among the thrombolytic agents, it is the least expensive but most commonly associated with adverse effects, including allergic reactions and hypotension.
Urokinase is also a plasminogen activator that is normally present in the urine. It is the major activator of fibrinolysis in the extravascular compartment, in contrast to tPA which is largely responsible for initiating intravascular fibrinolysis. Because the FDA-approved duration for tPA delivery is two hours, streptokinase and urokinase are rarely used today.]
What is the treatment for a hemophilia A patient with epistaxis, intracerebral hemorrhage, or hematuria?
Factor VIII concentrate of cryoprecipitate
[UpToDate: Serious or life-threatening bleeding in a patient with hemophilia is a medical emergency that requires prompt evaluation and immediate therapy with replacement factor. For patients with potentially serious or life-threatening bleeding, it is important to initiate treatment immediately, even before the diagnostic assessment is completed.
Serious or life-threatening bleeding includes any of the following:
- Bleeding in the central nervous system.
- Ocular bleeding.
- Bleeding in the hip.
- Deep muscle bleeding with neurovascular compromise or the potential for neurovascular complications.
- Intra-abdominal bleeding.
- Bleeding that could affect the airway (eg, into the throat or neck).
- Bleeding severe enough to result in anemia and potentially require red blood cell transfusion(s).
- Prolonged bleeding that is not adequately responding to home-based therapy.
- Iliopsoas bleeding.
- Significant injuries such as motor vehicle accidents or falls from distances of several feet or more.
An acutely hemorrhaging hemophilic patient should be transported to a facility equipped to handle the event that has the appropriate replacement products. Guidelines from the United Kingdom Haemophilia Centre Doctors Organization (UKHCDO) suggest that the maximum time between arrival to the hospital and clinical assessment should not exceed 15 minutes, and if treatment for bleeding is required, the maximum time to its delivery should not exceed 30 minutes. If the patient has the appropriate replacement therapy at home, this product may be administered before leaving or on route to the facility, as long as the bleeding is not life-threatening and this does not result in delays. In life-threatening circumstances, emergency medical transport should be called and the products should be administered on-route.
As noted above, factor administration should not be delayed while awaiting imaging studies in a patient with a concerning injury or suspected central nervous system bleeding. All significant head injuries must be considered nontrivial unless proven otherwise by observation and imaging (eg, with computed tomography [CT] or magnetic resonance imaging [MRI]). If there is doubt about the seriousness of bleeding, it is preferable to treat the patient as if the bleeding is serious (ie, “if in doubt, treat”). Further, the importance of urgently giving the factor infusion outweighs considerations of the specific factor preparation (ie, “give the appropriate product that is available rather than spending time trying to obtain a different product”).
Other hemostatic therapies for individuals with inhibitors or those whose bleeding is not controlled by factor infusion are presented below.
For severe bleeding, the factor activity level should be maintained above 50% at all times. An immediate dose of factor should be given to raise the peak factor level to 80% to 100%, and additional doses should be timed to occur when a factor activity level of approximately 50% is achieved, so the patient’s circulating factor level does not drop below 50%. Another option is to give a dose of factor to raise the level to 80% to 100%, followed by continuous infusion to maintain a consistent hemostatic level. Administration of factor should not be delayed while awaiting imaging studies.]

Which coagulation factor helps crosslink fibrin?
Factor XIII
[UpToDate: Activated factor XIII stabilizes and crosslinks overlapping fibrin strands.]

Which condition is diagnosed by a prolonged PTT that is not corrected by FFP, a positive Russell Viper venom time, and a false-positive RPR test for syphilis?
Anti-phospholipid antibody syndrome
[UpToDate: Antibody testing in patients with suspected APS involves immunoassays for antibodies to cardiolipin and beta2-glycoprotein (GP) I and a functional assay for the lupus anticoagulant (LA) phenomenon:
Anticardiolipin antibodies (aCL); immunoglobulin G (IgG) and/or IgM by enzyme-linked immunosorbent assay (ELISA).
Anti-beta2-GP I antibodies; IgG and/or IgM by ELISA.
LA testing is a three-step procedure:
- Demonstration of a prolonged phospholipid-dependent screening test of hemostasis. Commonly used screening tests include the dilute Russell viper venom time (dRVVT) and an activated partial thromboplastin time (aPTT) that has been optimized for this purpose (aPTT or lupus aPTT).
- Mixing patient plasma with normal plasma fails to correct the prolonged screening test(s). This eliminates the possibility that prolongation of the screening test is due to a coagulation factor deficiency. If the coagulation test remains prolonged after the addition of normal plasma, an inhibitor is present.
- Addition of excess phospholipid shortens or corrects the prolonged coagulation test (demonstration of phospholipid-dependence).
LA are characterized by correction of the prolonged clotting time with added phospholipid but not with control plasma, confirming that the coagulation inhibitor is phospholipid-dependent.
The above aPL testing is consistent with recommendations from the revised Sapporo classification criteria
A history of a false positive serologic test for syphilis may also be a clue to the presence of antiphospholipid antibodies (aPL). This phenomenon occurs because the antigen used in the Venereal Disease Research Laboratory (VDRL) and rapid plasma reagin (RPR) tests contains cardiolipin.]
Which type of von Willebrand’s disease causes the most severe bleeding?
Type III
[UpToDate: Von Willebrand factor (VWF) plays an important role in primary hemostasis by binding to both platelets and endothelial components, forming an adhesive bridge between platelets and vascular subendothelial structures at sites of endothelial injury and between adjacent platelets in areas with high shear. It also contributes to fibrin clot formation by acting as a carrier protein for factor VIII, which has a greatly shortened half-life and abnormally low concentration unless it is bound to VWF. Von Willebrand disease (VWD) is characterized by mutations that lead to a decrease in the level or impairment in the action of von Willebrand factor (VWF).
Type 1 VWD, an autosomal dominant disease, is the most common, accounting for approximately 75% of patients. The clinical presentation of type 1 VWD varies from mild to severe as determined by bleeding symptoms, but some individuals are asymptomatic and detected incidentally in studies investigating a relative for VWD. Type 1 VWD represents a partial quantitative deficiency of von Willebrand factor; many of the mutations remain undefined.
Type 2 VWD contains four subtypes in which VWF is qualitatively abnormal, as demonstrated by VWF multimer patterns, RIPA, and an abnormally low VWF activity to antigen ratio (types 2A, 2B, and 2M), or by other special assays such as a quantitative assay of the patient’s VWF binding capacity for factor VIII (type 2N). Type 2A accounts for approximately 10% to 15% of cases of VWD, and is usually transmitted as an autosomal dominant trait. Affected patients typically present with moderate to moderately severe bleeding. Type 2B VWD accounts for approximately 5% of cases of VWD, and is transmitted as an autosomal dominant trait. Affected patients generally present with moderate or moderately severe bleeding. The abnormal VWF in this disorder has a “gain of function”, binding more readily to the platelet receptor, glycoprotein Ib. The increase in binding of larger multimers to platelet GP Ib results in their loss from the circulation and, in some patients, thrombocytopenia occurs due to clearance or sequestration of the small platelet aggregates that are formed.
Type 3 VWD is a rare disease. Affected patients present with severe bleeding involving both the skin and mucous membrane surfaces (due to decreased VWF) and soft tissues and joints (due to the low concentration of factor VIII). Type 3 VWD is characterized by a marked decrease or absence of detectable VWF due to homozygous or compound heterozygous mutations, some of which result in loss of VWF mRNA expression.]

Which type of von Willebrand’s disease is characterized by a reduced quantity of vWF?
Type I
[Tx: recombinant Factor VIII and vWF, DDAVP, cryoprecipitate]
[UpToDate: Von Willebrand disease (VWD) is the most common inherited bleeding disorder, affecting up to 1% of the population as assessed by random laboratory screening, although only approximately 1% of these individuals are appreciably symptomatic. It is characterized by mutations that lead to a decrease in the level or impairment in the action of von Willebrand factor (VWF). Most cases are transmitted as an autosomal dominant trait that affects males and females equally. There are also acquired forms of VWD that are caused by several different pathophysiologic mechanisms.
Von Willebrand factor (VWF) plays an important role in primary hemostasis by binding to both platelets and endothelial components, forming an adhesive bridge between platelets and vascular subendothelial structures at sites of endothelial injury and between adjacent platelets in areas with high shear. It also contributes to fibrin clot formation by acting as a carrier protein for factor VIII, which has a greatly shortened half-life and abnormally low concentration unless it is bound to VWF.
Type 1 VWD, an autosomal dominant disease, is the most common, accounting for approximately 75% of patients. The clinical presentation of type 1 VWD varies from mild to severe as determined by bleeding symptoms, but some individuals are asymptomatic and detected incidentally in studies investigating a relative for VWD. Type 1 VWD represents a partial quantitative deficiency of von Willebrand factor; many of the mutations remain undefined.
Type 2 VWD contains four subtypes in which VWF is qualitatively abnormal, as demonstrated by VWF multimer patterns, RIPA, and an abnormally low VWF activity to antigen ratio (types 2A, 2B, and 2M), or by other special assays such as a quantitative assay of the patient’s VWF binding capacity for factor VIII (type 2N). Type 2A accounts for approximately 10% to 15% of cases of VWD, and is usually transmitted as an autosomal dominant trait. Affected patients typically present with moderate to moderately severe bleeding. Type 2B VWD accounts for approximately 5% of cases of VWD, and is transmitted as an autosomal dominant trait. Affected patients generally present with moderate or moderately severe bleeding. The abnormal VWF in this disorder has a “gain of function”, binding more readily to the platelet receptor, glycoprotein Ib. The increase in binding of larger multimers to platelet GP Ib results in their loss from the circulation and, in some patients, thrombocytopenia occurs due to clearance or sequestration of the small platelet aggregates that are formed.
Type 3 VWD is a rare disease. Affected patients present with severe bleeding involving both the skin and mucous membrane surfaces (due to decreased VWF) and soft tissues and joints (due to the low concentration of factor VIII). Type 3 VWD is characterized by a marked decrease or absence of detectable VWF due to homozygous or compound heterozygous mutations, some of which result in loss of VWF mRNA expression.]

Hemophilia A results from a deficiency in what?
Factor VIII
[UpToDate: The factor VIII gene is one of the largest genes known, comprising about 0.1% of the X chromosome. The gene that encodes factor VIII is divided into 26 exons that span 186,000 base pairs. Factor VIII contains several areas of internal homology, consisting of a heavy chain with A1 and A2 domains; a connecting region with a B domain; and a light chain with A3, C1, and C2 domains.
Some of these domains have specific functions. For example, different epitopes on the C2 domain are responsible for binding to the procoagulant phospholipid phosphatidylserine on activated platelets and endothelial cells, von Willebrand factor (which importantly slows the catabolism of factor VIII), factor Xa, and thrombin. Two domains contribute to the binding of factor IXa (A2 domain and the A1/A3-C1-C2 dimer).
Hemophilia A genes — Examination of hemophilic genes has not demonstrated a uniform abnormality. Instead, numerous different mutations in the factor VIII gene have been described.]
What is the treatment for Factor VII deficiency?
Recombinant factor VII concentrate or FFP
[UpToDate: Bleeding can be managed with recombinant human factor VII in the activated form (rFVIIa; NovoSeven RT, Niastase, Niastase RT), which became available in 1999; or factor VII concentrates, which are available in some European countries.
Recommended dosing of rFVIIa is 15 to 30 mcg/kg every 12 hours, and dosing of factor VII concentrates is 30 to 40 international units/kg, repeated every 6 to 12 hours, with the goal of maintaining factor VII activity levels above 15% to 20%. Higher doses may be required in severe or life-threatening bleeding.]

In addition to thrombocytopenia, what can heparin-induced thrombocytopenia (HIT) cause?
Platelet aggregation and thrombosis
[Forms a white clot]
[UpToDate: HIT results from an autoantibody directed against endogenous platelet factor 4 (PF4) in complex with heparin. This antibody activates platelets and can cause catastrophic arterial and venous thrombosis with a mortality rate as high as 20%; although, more recently with improved recognition and early intervention, these rates have been reported as below 2%. In those suspected of having HIT based on clinical grounds, all exposure to heparin should be eliminated immediately and a non-heparin anticoagulant should be administered until a complete diagnosis can be made.]

Which coagulation factor gets activated during cardiopulmonary bypass, resulting in a hypercoagulable state?
Factor XII (Hageman factor)
What is deficient in Bernard Soulier syndrome?
GpIb receptor on platelets is deficient resulting in platelets being unable to bind collagen

What is deficient in Glanzmann’s thrombocytopenia?
GpIIb/IIIa receptor on platelets is deficient resulting in platelets being unable to bind to each other
[UpToDate: Glanzmann thrombasthenia is an autosomal recessive bleeding disorder characterized by a defect in the platelet integrin αIIbβ3 (integrin alphaIIbbeta3; previously known as GPIIb/IIIa); clinical manifestations are limited to bleeding, which is mostly mucocutaneous. The presence of mucocutaneous bleeding and a normal platelet count but with single isolated platelets without any platelet clumping on examination of a non-anticoagulated peripheral blood smear should raise the possibility of this disorder. Platelet aggregometry is distinctly abnormal.
This disorder may also occur in combination with defects in leukocyte function in the disorder leukocyte adhesion deficiency III, and should be suspected in infants with concomitant leukocytosis, delayed separation of the umbilical cord, or severe bacterial infections.
Antibodies to integrin αIIbβ3 and/or HLA antigens may occur in subjects with Glanzmann thrombasthenia who have received multiple platelet transfusions, resulting in refractoriness to such transfusions.
The use of recombinant factor VIIa and other hemostatic agents in such settings has been helpful in controlling bleeding, although controlled efficacy studies are lacking.]

How does aspirin prolong bleeding time?
It inhibits cyclooxygenase in platelets, decreasing levels of TXA2
[UpToDate: Irreversibly inhibits cyclooxygenase-1 and 2 (COX-1 and 2) enzymes, via acetylation, which results in decreased formation of prostaglandin precursors; irreversibly inhibits formation of prostaglandin derivative, thromboxane A2, via acetylation of platelet cyclooxygenase, thus inhibiting platelet aggregation; has antipyretic, analgesic, and anti-inflammatory properties.]

Which patients are especially susceptible to Warfarin-induced skin necrosis?
Patients with relative protein C deficiency
[UpToDate: Warfarin-induced skin necrosis is a complication of warfarin therapy in which the patient develops demarcated areas of purpura and necrosis due to vascular occlusion. The appearance may be similar to that of neonatal purpura fulminans and may affect one or more areas of skin including the extremities, breasts, trunk, or penis.
The mechanism of warfarin-induced skin necrosis involves a transient hypercoagulable state during initial warfarin administration that in turn leads to vascular occlusion and tissue infarction followed by extravasation of blood.
The half-lives vary among the vitamin K-dependent coagulation factors (factors II, VII, IX, and X) and natural anticoagulants (protein S and protein C), and as a result, the factors with the shorter half-lives (half-lives for factor VII and protein C of 8 and 14 hours, respectively) are depleted more rapidly than the others. Laboratory studies of thrombin generation using an assay for the activation of prothrombin using the generation of fragment F1+2 have suggested that effects on protein C (ie, a procoagulant effect) predominate over effects on factor VII in vivo.
The skin lesions in warfarin-induced skin necrosis typically form during the first few days of warfarin therapy, often in the setting of large loading doses of 10 or more milligrams of warfarin per day. If the patient is receiving heparin and warfarin therapy, the lesions may appear upon discontinuation of the heparin. The lesions typically marginate over a period of hours from an initial central erythematous macule, similar to neonatal purpura fulminans. If a product containing protein C is not rapidly administered, the affected cutaneous areas become edematous, develop central purpuric zones, and ultimately become necrotic. Biopsy of the lesions is not generally performed, but if a biopsy is obtained it may show diffuse microthrombi within dermal and subcutaneous capillaries, venules, and deep veins, with endothelial cell damage, resulting in ischemic skin necrosis and marked red blood cell extravasation. These findings are indistinguishable from other thrombotic skin lesions including antiphospholipid syndrome (APS), disseminated intravascular coagulation (DIC), and heparin-induced thrombocytopenia (HIT).
The incidence of warfarin-induced skin necrosis in individuals with protein C deficiency is unknown, as most descriptions are in the form of case reports. Warfarin-induced skin necrosis is not pathognomonic for protein C deficiency; it has been described in individuals with other inherited thrombophilias (factor V Leiden mutation, protein S deficiency) and transient reductions of protein C levels (eg, in the setting of cancer).]
What is the half-life of Bivalrudin?
25 minutes
[Metabolized by proteinase enzymes in the blood]
[UpToDate:
- Normal renal function (CrCl ≥90 mL/minute): 25 minutes
- Severe renal impairment (CrCl 10 to 29 mL/minute): 57 minutes
- Dialysis-dependent patients (off dialysis): 3.5 hours]
Which system in the body clears Heparin?
The reticuloendothelial system
[UpToDate: Heparin is metabolized by the liver and may be partially metabolized in the reticuloendothelial system . It is excreted in the urine (small amounts as unchanged drug). At therapeutic doses, elimination occurs rapidly via nonrenal mechanisms. With very high doses, renal elimination may play more of a role; however, dosage adjustment remains unnecessary for patients with renal impairment.
LMW heparins are primarily excreted by the kidney, so their biological half-life may be prolonged in patients with renal failure. Uremia may also contribute to increased bleeding risk. As a result, most trials have excluded patients with creatinine clearance (CrCl) ≤30 mL/min. In a systematic review and meta-analysis of studies that evaluated bleeding risk in individuals with renal insufficiency who were receiving a LMW heparin, patients with a CrCl ≤30 mL/min receiving LMW heparin were more likely to have bleeding than those with a CrCl >30 mL/min, (odds ratio [OR] 2.25; 95% CI 1.19-4.27). Individuals with CrCl ≤30 mL/min who were receiving enoxaparin at therapeutic doses had higher levels of anti-factor Xa activity compared with individuals without renal insufficiency or those who had dose adjustments based on renal function or anti-factor Xa activity, although anti-factor Xa activity measurements in patients on LMW heparin have not been correlated with clinical events. In contrast, tinzaparin and dalteparin do not appear to bioaccumulate in individuals with this degree of renal insufficiency, although less rigorous evidence is available for these products.
Options for management depend on the degree of renal insufficiency and the available LMW heparin. For those with a CrCl ≤30 mL/min, use of unfractionated heparin avoids the problems associated with impaired renal clearance of LMW heparin. If LMW heparin is used in an individual with renal insufficiency, dose-reduction and/or adjustment based on anti-factor Xa levels may be appropriate, especially for enoxaparin.]
What is the appropriate treatment for a patient with 2 or more prior DVTs or a significant PE who develops a postoperative DVT?
Lifetime Warfarin
[UpToDate: Most patients with a first episode of venous thromboembolism (VTE; proximal deep venous thrombosis [DVT] and/or pulmonary embolus [PE]) are anticoagulated for a finite period of 3 to 12 months. Select patients benefit from indefinite anticoagulation which is administered with the primary goal of reducing the lifetime risk of recurrent thrombosis and VTE-associated death.
The decision to anticoagulate indefinitely should be individualized and based upon an estimate of the risk of recurrence and bleeding in the context of the patient’s values and preferences. In general, the following applies.
For most patients with a first episode of unprovoked proximal DVT, unprovoked symptomatic PE, or active cancer in whom the risk of bleeding is low to moderate, we suggest indefinite anticoagulation rather than stopping therapy after 3 to 12 months (Grade 2B). In patients with a recurrent episode of unprovoked VTE, we recommend indefinite anticoagulation rather than stopping therapy after 3 to 12 months (Grade 1B).
Indefinite anticoagulation should not be routinely administered to patients with a provoked episode of VTE with major transient risk factors (eg, surgery, cessation of hormonal therapy) (Grade 1B). We also avoid indefinite anticoagulation in those with a high bleeding risk; however, should the risk for bleeding resolve, indefinite anticoagulation may be reconsidered.
For most patients with recurrent provoked VTE or a first episode of provoked VTE with irreversible, multiple, or minor risk factors, a first episode of unprovoked isolated distal DVT or an unprovoked episode of incidental PE, therapy must be individualized based upon a careful assessment of patient-specific risks of bleeding and thrombosis. There are wide variations in both the recurrence risk and benefit in these populations.]

What platelet concentration do we want before and after surgery?
- Greater than 50,000 before surgery
- Greater than 20,000 after surgery
[UpToDate: The concept of a “safe” platelet count is imprecise, lacks evidence-based recommendations, and depends on the disorder and on the patient (even with the same disorder). The following may be used as guides, but should not substitute for clinical judgment based on individual patient and disease factors:
- Surgical bleeding generally may be a concern with platelet counts <50,000/microL (<100,000/microL for some high-risk procedures such as neurosurgery or major cardiac or orthopedic surgery).
- Severe spontaneous bleeding is most likely with platelet counts <20,000 to 30,000/microL, especially below 10,000/microL.
It is also important to consider other factors that may affect bleeding risk (eg, platelet function defects, coagulation abnormalities). When present, these factors may contribute to bleeding risk and may be more concerning than the low platelet count.]
What is the treatment for uremic platelet dysfunction?
Hemodialysis
[DDAVP and platelets can be given if this is not fully effective]
[UpToDate: Patients who are actively bleeding or who are about to undergo a surgical procedure should have correction of platelet dysfunction. Treatment options include correction of anemia, desmopressin (dDAVP), dialysis, estrogens, or cryoprecipitate. Therapies vary in their onset and duration of action, and most have been shown only to reduce the bleeding time or in vitro tests of platelet function rather than to reduce active bleeding or the risk of bleeding with invasive procedures.
Raising the hemoglobin to approximately 10 g/dL may reduce the bleeding time. The improvement in platelet function will persist for as long as the hemoglobin remains elevated. Erythropoietic-stimulating agents (ESAs) may also have a direct beneficial effect on platelet function.
Desmopressin provides the simplest and most rapid acute treatment for platelet dysfunction in the uremic patient. Administration of desmopressin at a dose of 0.3 mcg/kg given in 50 mL of saline over 15 to 30 minutes intravenously or by subcutaneous injection is preferred; a dose of 3 mcg/kg can also be given intranasally. The improvement in bleeding time begins within 1 hour and lasts 4 to 8 hours. The response to subsequent doses is generally diminished (tachyphylaxis).
Either hemodialysis or peritoneal dialysis can partially correct the bleeding time in approximately two-thirds of uremic patients. Hemodialysis should be performed without systemic anticoagulation.
Prolonged control of bleeding may be achieved by the administration of conjugated estrogens (0.6 mg/kg intravenously daily for five days, 2.5 to 25 mg orally per day, or 50 to 100 mcg of transdermal estradiol twice weekly). These agents begin to act on the first day, with peak control reached over five to seven days; the duration of action is 1 week or more after therapy has been discontinued.
The infusion of cryoprecipitate (10 units intravenously every 12 to 24 hours) can shorten the bleeding time in many uremic patients. The improvement in bleeding time begins within 1 hour and lasts 4 to 24 hours. Potential infectious complications limit the use of cryoprecipitate to patients with life-threatening bleeding who are resistant to treatment with desmopressin and blood transfusions.]
What is the inheritance pattern of hemophilia B?
Sex-linked recessive
[UpToDate: Factor VIII deficiency (hemophilia A) affects 1 in 5000 to 10,000 males; roughly 60% have severe disease, with factor VIII activity less than 1% of normal.
Factor IX deficiency (hemophilia B) affects 1 in 25,000 to 30,000 males; approximately one-half have mild to moderate disease, with factor IX activity greater than 1% of normal.
Severe factor VIII or factor IX deficiency leads to bleeding because of the role these factors play in the intrinsic pathway X-ase (ten-ase). The X-ase complex consists of activated factor IX (factor IXa) as the protease; activated factor VIII (factor VIIIa), calcium, and phospholipids as the cofactors; and factor X as the substrate.
Hemophilia A and B are X-linked recessive disorders, which explains who is likely to bleed and the modes of genetic transmission. These hemophilias occur almost exclusively in a male having one defective copy of the relevant gene on his X chromosome (ie, he is hemizygous for the defect). Because the affected male will transmit a normal Y chromosome to all his sons and an abnormal X chromosome to all his daughters, his sons will not be affected and all of his daughters will be carriers (ie, they are heterozygous for the defect).]

What is the normal half-life of PMNs?
1-2 days
What is the treatment for Bernard Soulier syndrome?
Platelets
[UpToDate: Inherited platelet disorders with giant platelets are quite rare. These include platelet glycoprotein abnormalities (eg, Bernard-Soulier syndrome), deficiency of platelet alpha granules (eg, gray platelet syndrome), the May-Hegglin anomaly, which also involves the presence of abnormal neutrophil inclusions (ie, Döhle-like bodies), and some kindreds with type 2B von Willebrand disease (the Montreal platelet syndrome).
Patients with these disorders who have bleeding diatheses are usually treated with platelet transfusions. In a small study in subjects with MYH9-RD and platelet counts <50,000/microL, treatment with the non-peptide thrombopoietin receptor agonist eltrombopag resulted in major responses (ie, platelet counts of at least 100,000/microL or three times baseline) in 8 of the 12 so treated. Bleeding tendency disappeared in 8 of the 10 subjects with bleeding symptoms at baseline.]