Pulm/Renal - Biochemistry - The Golgi Apparatus: Mucolipidoses; Glycobiology: Mucopolysaccharidoses & Sphingolipidoses Flashcards
Describe the hypothesized signalling mechanism by which proteins that are being synthesized end up in the rough endoplasmic reticulum.
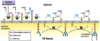
(1) Translation begins in the cytosol; an RER-targeting sequence on the partially synthesized protein is bound by a signal recognition particle (SRP).
(2) Upon binding to an SRP receptor in the RER, GTP hydrolysis opens an associated translocon protein (the RER channel opens, and the SRP dissociates from the ribosome).
(3) Translation continues directly through the translocon and into the RER lumen; signal peptidase cleaves the signalling sequence on the synthesized protein.

In the signal hypothesis of RER synthesis, what binds to signalling sequences on partially synthesized proteins?
Where are these proteins when this occurs?
Where are they taken next?
Signal receptor particles (SRPs);
the cytosol;
SRP receptors on the RER

Secretory protein synthesis begins in the cytosol. How are these proteins targeted to the RER?
What effect does this targeting have on translation?
By signal receptor proteins (SRPs) that bind SRP receptors on the RER surface;
translation is blocked when SRPs bind the partially synthesized proteins, but it is restored when the SRP dissociates and the ribosome is bound to the SRP receptor

What must occur for both signal receptor proteins to dissociate from SRP receptors on the RER and also the associated translocons to open?
GTP hydrolysis

Secretory proteins begin their translation in the ________, where an N’ signalling sequence targets them towards the RER.
Upon entering the RER, what cleaves this signalling sequence from the newly synthesized proteins?
Cytosol;
signal peptidase (in the RER membrane)

Signal receptor particles (SRPs) bind what to bring partially synthesized secretory proteins to the RER?
An N’ signalling sequence on the partially synthesized protein and the 60s ribosomal subunit

What are the two main purposes of protein glycosylation?
What are some other purposes?
Folding, targeting;
E-leaflet protein stability, enhanced cell-to-cell contact
________ are carbohydrate-binding proteins, and can interact with other glycosylated proteins.
Lectins
Lectins are ___________-binding proteins, and can interact with other ___________ proteins.
Carbohydrate;
glycosylated
True/False.
Glycosylation is typically the addition of single glucose monomers to amino acid side chains.
False;
it is usually the addition of glycans (either single oligosaccharides or complexes) to amino acid side chains.
What are the two types of protein glycosylation?
O-linked;
N-linked
N-linked glycosylation occurs in the _____________.
O-linked glycosylation occurs in the _____________.
RER;
Golgi apparatus
Describe O-linked glycosylation (in terms of starting substrate and any involved amino acids or enzymes).
Give an example.
Individual carbohydrates are sequentially added to –OH groups of serine or threonine by glycosyl transferases;
the formation of ABO blood antigens
Describe N-linked glycosylation (in terms of starting substrate and any involved amino acids or enzymes).
What amino acid sequence is required?
Preformed oligosaccharide complexes are added to asparagine residues as the peptide is translocated through the RER membrane (translocation process shown in attached image);

Asn - X - (Ser/Thr)
__-linked glycosylation is characterized by the addition of preformed oligosaccharide complexes to __________ residues.
__-linked glycosylation is characterized by the sequential addition of individual carbohydrates to ___________ residues.
N, Asparagine (Asn-X(Ser/Thr);
O, Serine/Threonine
Describe the steps of synthesis of the oligosaccharide complex used in N-linked glycosylation.
(1) UDP-linked sugars are added to dolichol phosphate from the cytosolic side.
(2) This complex then ‘flips’ from the cyotosolic to the RER lumen side.
(3) The completed oligosaccharide complex can then be cleaved and added to asparagine residues by oligosaccharyl transferase.

In synthesis of the oligosaccharide complexes used in N-linked glycosylation, _______-linked sugars are first added to _____________ _____________ on the cytosolic side of the RER membrane.
UDP;

dolichol phosphate
What must occur for the oligosaccharide complexes used in N-linked glycosylation to enter the RER lumen?
It ‘flips’ from the cytosolic side to the RER lumen

What is the first substrate for synthesis of the oligosaccharide complexes involved in N-linked glycosylation?
It is attached to what substance in the RER membrane?
What substance can be used in the lab to inhibit this process, thus disrupting proper protein folding?
UDP-GluNAc;
dolichol phosphate;
tunicamycin

N-linked glycosylation is accomplished via what enzyme?
Oligosaccharyl transferase

What effect does tunicamycin have on cells?
What is its clinical use?
Inhibition of first step of N-linked glycosylation
–> inhibition of proper protein folding in eukaryotic cells;
none (laboratory use only)
True/False.
Disulfide bonds form in many proteins synthesized both in the cytosol and the RER.
False.
Disulfide bonds form mainly in secretory proteins and proteins of the E-leaflet;
they are only made in RER-synthesized proteins
Why do disulfide bonds only form in the RER lumen?
Via what enzyme?
It is an oxidative environment;
protein disulfide isomerase
What is the most common genetic cause of liver disease (jaundice, cirrhosis) in children?
What is the most common disorder necessitating pediatric liver transplants?
α-1 antitrypsin deficiency;
α-1 antitrypsin deficiency
What percentage of emphysema cases are due to an α-1 antitrypsin deficiency?
1 - 3%
What enzyme(s) are inhibited by α-1 antitrypsin?
From what organ(s) is this protein secreted?
Elastase, trypsin;
the liver
What is the main molecular cause of an α-1 antitrypsin deficiency?
What tissues does this damage?
A point mutation inhibiting proper folding of the α-1 antitrypsin protein in the RER;
pulmonary (increased elastase activity),
hepatic (α-1 antitrypsin crystalline aggregate buildups)
GI (increased trypsin activity)
What cell type(s) secrete elastase?
Neutrophils
How does an α-1 antitrypsin deficiency damage the lungs?
How does an α-1 antitrypsin deficiency damage the GI tract?
How does an α-1 antitrypsin deficiency damage the liver?
Decreased elastase inhibition;
decreased trypsin inhibition;
misfolded α-1 antitrypsin aggregates
What are the signs/symptoms of an α-1 antitrypsin deficiency?
[Pulmonary] Shortness of breath, fatigue;
[hepatic] jaundice, cirrhosis;
[GI] bleeding
What are the two main pillars of α-1 antitrypsin deficiency treatment?
Mainly the following:
(1) prophylaxis against pulmonary issues (e.g. vaccinations, bronchodilators),
(2) liver transplants
(Note: α-1 antitrypsin IV infusions may or may not be beneficial.)
What are the three primary locations to which the Golgi sends synthesized proteins?
(1) Secretory vesicles;
(2) endosomes/lysosomes;
(3) the plasma membrane
What are the three main sections (cisternae) of the Golgi apparatus?
The cis-Golgi, medial-Golgi, and trans-Golgi
What are the two forms of substrate movement through the Golgi apparatus?
Vesicular (anterograde, retrograde);
cisternal maturation (cis to medial to trans)
The cisternae of the Golgi apparatus facing the RER is the ___-Golgi.
The cisternae of the Golgi apparatus between the other two cisternae is the ___-Golgi.
The cisternae of the Golgi apparatus facing the plasma membrane is the ___-Golgi.
- Cis*;
- medial*;
- trans*

What protein is involved in anterograde Golgi budding and vesicular movement?
What protein is involved in retrograde Golgi budding and vesicular movement?
COP-II (RER to cis-Golgi);
COP-I (cis-Golgi to RER)
What is an important reason why retrograde Golgi transport is needed?
As cisternal maturation occurs (cis- turning into medial- turning into trans-Golgi), the necessary proteins are shuttled back from the maturing cisternae to the cisternae before them

The COP-I protein in the Golgi apparatus facilitates what event?
The COP-II protein in the Golgi apparatus facilitates what event?
Retrograde vesicular transport
either from the cis-Golgi to the ER
or from cisternae to earlier cisternae;
anterograde vesicular transport from the ER to the cis-Golgi only

What protein mediates vesicular transport from the Golgi apparatus out to some other location (e.g. endosomes, lysosomes, melanosomes, platelet vesicles)?
Clathrin
What is the role of coat proteins in the Golgi apparatus?
What are some clathrin-associated examples?
To form the curvature of transport vesicles;
AP1, AP2, AP3
What GTPase is associated with the COP-I protein in Golgi vesicular transport?
What GTPase is associated with the COP-II protein in Golgi vesicular transport?
What GTPase is associated with clathrin in Golgi vesicular transport?
ARF;
SAR1;
ARF
What proteins allow for vesicular interactions with target locations?
SNARE proteins

- (v-SNARE = vesicular SNARE;*
- t-SNARE = target SNARE)*
What are some of the major players involved in vesicular transport formation, coating, de-coating, docking, etc.?
Coat proteins;
GTPases (ARF or SAR1);
secretory product receptors;
SNARE proteins

True/False.
GTPases are important in Golgi vesicular coating when bound to GTP, and they are important in vesicular decoating after hydrolyzing that GTP to GDP.
True.

Where does high mannose glycosylation occur (addition of GlcNAc and mannose to synthesized cellular products)?
Where does complex glycosylation occur (addition of galactose and neuraminic acid to synthesized cellular products)?
The cis-Golgi;
the trans-Golgi

What sugars can be added to a product in the cis-Golgi?
What sugars can be added to a product in the medial-Golgi?
What sugars can be added to a product in the trans-Golgi?
Mannose + GlcNAc
Fucose + Mannose + GlcNAc
Galactose + N-acetylneuraminic acid

What signalling sugar targets Golgi products to the lysosome?
What vesicle type will be used (COP-I, COP-II, or clathrin)?
What coat proteins are involved?
What GTPase is involved?
Mannose-6-phosphate;
clathrin;
AP3;
ARF
Coat protein AP_ aids in delivery of Golgi products to the lysosomes.
Coat protein AP_ aids in delivery of Golgi products to the endosomes.
Coat protein AP_ is involved in receptor-mediated endocytosis.
3;
1;
2
Golgi products lacking mannose-6-phosphate are destined for ____________.
Golgi products with attached mannose-6-phosphate go to the ____________.
Secretion;
lysosomes
What is the molecular cause of I-Cell disease?
Failure of mannose-6-phosphate addition to lyosomal proteins
(lysosomal proteins secreted)
What disease results as a failure of mannose-6-phosphate addition to lysosomal proteins?
What effect does this have on cellular activity?
I-Cell disease;
buildup of material-filled endosomes with no acid hydrolases to break any of it down
(hence, I-Cell disease means inclusion-Cell disease)

What are some of the signs and symptoms of I-Cell disease?
When does it manifest?
Coarse facial features and craniofacial abnormalities,
low birth weight and slowed growth rate,
psychomotor retardation;
at birth

What enzyme is blocked in I-Cell disease?
GlcNAc phosphotransferase
(normally, this adds the phosphate with GlcNAc to mannose chains and then cleaves the GlcNAc to leave just the phosphate)

How is I-Cell disease different from mucopolysaccharidoses or sphingolipidoses?
Instead of being an enzyme deficiency that prevents breakdown of a certain sugar (i.e. a mucopolysaccharidosis) or a certain fat (i.e. a sphingolipidosis),
there is an inability to break down any product brought into the cell for digestion
What is a mucopolysaccharidosis?
(examples?)
What is a sphingolipidosis?
(examples?)
What is a mucolipidosis?
(examples?)
Enzyme deficiency –> inability to breakdown a specific sugar
(e.g. Hurler, Hunter, or Sanfilippo syndromes)
Enzyme deficiency –> inability to breakdown a specific lipid
(e.g. Niemann-Pick, Fabry, Krabbe, Gaucher, or Tay-Sachs, or metachromatic leukodystrophy syndromes)
A disease with features of both of the above categories
(e.g. I-Cell disease, pseudo-Hurler syndrome, or sialidase deficiency)
What are some example mucopolysaccharidoses?
What is the general defect in these disorders?
Hurler, Hunter, or Sanfilippo syndromes;
an enzyme deficiency leads to an inability to breakdown a specific sugar
What are some example sphingolipidoses?
What is the general defect in these disorders?
Niemann-Pick, Fabry, Krabbe, Gaucher, or Tay-Sachs, or metachromatic leukodystrophy syndromes
an enzyme deficiency leads to an inability to breakdown a specific lipid
What are some example mucolipidoses?
What is the general defect in these disorders?
I-Cell disease (ML 2), pseudo-Hurler polydystrophy (ML 3), sialidase deficiency (ML 1);
a disease with features of both a mucopolysaccharidosis and a sphingolipidosis (errors in both fat and sugar breakdown)
What enzyme is deficient in sialidosis (mucolipidosis type I) disease?
What enzyme is deficient in I-Cell (mucolipidosis type II) disease?
What enzyme is deficient in pseudo-Hurler (mucolipidosis type III) disease?
Sialidase
GlcNAc phosphotransferase (leads to exocytosis of all lysosomal enzymes)
GlcNAc phosphotransferase (though less severe than I-Cell disease)
What two mucolipidoses occur as a result of a deficiency in the GlcNAc phosphotransferase enzyme?
What is the result?
I-Cell disease (ML 2),
pseudo-Hurler polydystrophy (ML 3);
a lack of all lysosomal enzymes in the lysosome
How does an endosome become a lysosome?
It must be acidified (proton pump –> pH 5.0 - 6.0) and gain lysosomal enzymes (M6P –> acid hydrolases)
What structure becomes a lysosome?
What two structures can fuse with this structure to form lysosomes?
Endosome (early, then late);
phagosomes, autophagosomes

Endocytosis is different from phagocytosis in that endocytosis is an __vagination of the membrane while phagocytosis is an __vagination of the membrane.
in;
e

What substrate types can be broken down by lysosomal acid hydrolases?
Just about anything
(proteases, lipases, glycosidases, nucleases, sulfatases, phosphatases)
When does cholesterol dissociate from LDL upon being taken up by LDL receptors?
When does iron dissociate from apotransferrin upon being taken up by ferrotransferrin receptors?
The acidity of the lysosome causes them to dissociate;
the acidity of the lysosome turns Fe3+ into Fe2+, and it dissociates from apotransferrnin
What protein system normally allows for formation of vesicles in the lysosome but can be hijacked by HIV as a means of making extracellular vesicles for HIV ‘budding’ and spread?
The HRS-ESCRT system is hijacked by mono-ubiquinated HIV Gag proteins

Why is the HIV ‘Gag’ protein a target for HIV therapies?
Inhibiting it prevents HIV budding and spread

Are sugars in the human body typically in the D- or L-configuration?
What does it mean for them to be α or β?
D
(alcohol group ‘up’ at carbon 5, hydrogen ‘down’ at carbon 5);
whether the carbon 1 hydroxyl is ‘up’ (β) or ‘down’ (α)
(Note: the above α/β rule is correct for D-sugars. It is reversed for L-sugars.)

Mannose is the __ epimer of glucose.
Galactose is the __ epimer of glucose.
C2;
C4

Describe the Hayworth projection for glucose (α or β).

How can this D-glucose monomer be changed into N-acetylglucosamine?


How can this D-glucose monomer be changed into N-glucuronic acid?


What is the C2 epimer for D-glucose?
Mannose
What is the C4 epimer for D-glucose?
Galactose
Glucuronic acid is a(n) __-sugar.
Iduronic acid is a(n) __-sugar.
D;
L

What is another name for neuraminidase?
What is another name for N-acetylneuraminic acid?
Sialidase;
sialic acid
Glucose is a 6-carbon sugar.
N-acetylneuraminic (sialic) acid is a __-carbon sugar.
9
Lactose is a disaccharide made of what two monosacharrides connected by what type of linkage?
Glucose + galactose;
1,4 β-galactosidic (glycoside) linkage
True/False.
In order to combine glucose and galactose to make lactose, the glucose must be activated (often UDP-bound).
False.
In order to combine glucose and galactose to make lactose, the galactose must be activated (often UDP-bound).
When is lactose made in the body?
Only in females during lactation
True/False.
In order for monosacharrides to be added together, one of the sugars is usually nucleotide-activated (e.g. UDP-bound).
True.
Typically, UDP-galactose is added to N-acetylglucosamine to form N-acetyllactosamine.
During lactation, what protein do the mammary glands produce to alter the N-acetylglucosamine back to D-glucose (thus producing lactose in this reaction)?
What is the enzyme accomplishes both of these potential reactions?
Lactalbumin;
β-galactosyltransferase
In non-lactating mammary glands, UDP-galactose is added to ______________ to form ________________.
During lactation, lactalbumin is secreted for what purpose?
What is the enzyme accomplishes both of these potential reactions?
N-acetylglucosamine, N-acetyllactosamine;
to convert the N-acetylglucosamine to glucose;
β-galactosyltransferase
Lactalbumin is produced during lactation so the mammary glands produce ___________ instead of __________.
Lactose;
N-acetyllactosamine
What is another term for lactase?
β-galactosidase
O-linked glycosylation involves sequential addition of sugars to ___________ or ____________ amino acid residues.
N-linked glycosylation involves addition of a sugar complex to ____________ amino acid residues.
Serine, threonine;
asparagine
O-linked glycosylation involves sequential addition of ________ to serine or threonine amino acid residues.
N-linked glycosylation involves addition of _____________ (multiple words) to asparagine amino acid residues.
Monosacharrides;
preformed sugar complexs
__-linked glycosylation involves addition of a sugar complex to asparagine amino acid residues.
__-linked glycosylation involves sequential addition of monosacharrides to threonine or serine amino acid residues.
N;
O
Which is the acidic product of glycosylation, high mannose type or complex type?
This is due to the presence of what acid?
Complex type;
neuraminic (sialic) acid
Congenital disorders of glycosylation involve defects in the addition of __-glycans.
N
In general terms, what are type I congenital disorders of glycosylation?
And type II congenital disorders of glycosylation?
Inability to properly synthesize N-glycans or transfer them to a substrate;
inability to process the protein-bound oligsacharride chain
Which type of congenital disorder of glycosylation involves a failure of processing protein-bound N-glycans?
Which type of congenital disorder of glycosylation involves either a failure of synthesis of N-glycans or also a failure of transfer of the N-glycans to a protein substrate?
Type II;
type I
The adult form of hemoglobin is Hb__.
When this structure is glycosylated, it forms Hb___.
HbA;
HbA1C
(most stable form of glycosylated hemoglobin)
Which globin chain is glycosylated to form HbA1C?
The hemoglobin A β-globin chains
What enzyme is responsible for hemoglobin A glycosylation into HbA1C?
None
(this is non-enzymatic glycosylation)
What is a normal HbA1C level?
≤ 5.6%
What are the two main proteins found on the influenza virus surface?
Hemagglutinin (binds sialic/neuraminic acid)
neuraminidase (cleaves sialic/neuraminic acid)
What is the term for a protein that has a strong affinity for a sugar?
What is an example found on the influenza virus?
A lectin;
hemagglutinin
Influenza classifications and vaccine production typically revolves around what two proteins?
Hemagglutinin and neuraminidase
(e.g. H1N1 virus)
What influenza virus protein binds the sialic acid on our cell surfaces?
What influenza virus protein cleaves the sialic acid to release the virus?
Hemagglutinin;
neuraminidase
(sialic acid is also known as N-acetylneuraminic acid)
The H7N9 influenza virus refers to variations in what?
Versions of the proteins Hemagglutinin and Neuraminidase on the viral surface
What do the flu treatments Tamiflu (Oseltamivir) and Relenza (Zanamivir) do?
Block neuraminidase
(thus trapping newly formed flu viruses on the host cell surface)
Changes in influenza proteins hemagglutinin and neuraminidase occur each year due to ____________ _________.
Antigenic drift
True/False.
Glycosaminoglycans and mucopolysaccharides are synonyms.
True.
Hyaluronic acid, chondroitin sulfate, dermatan sulfate, keratan sulfate, heparin, and heparan sulfate are all examples of:
Glycosaminoglycans
(mucopolysaccharides)
Mucopolysaccharidoses are disorders caused by improper catabolism of what type of substrate?
Glycosaminoglycans
(mucopolysaccharides)
With the exception of hyaluronic acid, glycosaminoglycans (mucopolysaccharides) are always linked to ____ ________ to form ____________.
Core proteins;
proteoglycans
What is the basic structure of a mucopolysaccharide (glycosaminoglycan)?
A repeating disacharride
(1 hexose (or hexuronic acid) + 1 hexosamine)
Name a few example mucopolysaccharides (glycosaminoglycans).
Hyaluronic acid
Chondroitin sulfate
Dermatan sulfate
Keratan sulfate
Heparin
Heparan sulfate
What complex is made of many proteoglycans (core proteins + GAGs) bound to a single hyaluronic acid (another GAG)?
Aggrecan

Why is aggrecan important to normal ECM function?

It extremely hygroscopic (absorbs water like a sponge) and thus a great shock absorber
What are some general signs/symptoms of mucopolysaccharidoses?
Flat nasal bridge
Thick lips
Short stature
Intellectual disabilities
Organ enlargement (heart, liver, spleen)
Hunter syndrome is caused by a deficiency of what enzyme?
What does this enzyme do?
Iduronate sulfatase;
cleaves the sulfur group off dermatan sulfate and heparan sulfate so they can be broken down

Hurler syndrome is caused by a deficiency of what enzyme?
What does this enzyme do?
α-L-Iduronidase;
cleaves L-iduronic acid monomers off dermatan sulfate and heparan sulfate

What disorder results from an inability to remove the initial sulfur group from heparan (or dermatan) sulfate?
What disorder results from an inability to remove the iduronic acid from heparan (or dermatan) sulfate following the sulfur removal in the step above?
What disorder results from an inability to remove the sulfur group from the aminoglycan from heparan sulfate following iduronic acid removal in the step above?
Hunter syndrome
Hurler syndrome
Sanfilippo syndrome A

Hunter syndrome involves a deficiency of what enzyme?
This impairs removal of a sulfur from what glycosaminoglycan(s) (mucopolysaccharide(s))?
Iduronate sulfatase;
heparan sulfate, dermatan sulfate
Hurler syndrome involves a deficiency of what enzyme?
This impairs removal of what acid from what glycosaminoglycan(s) (mucopolysaccharide(s))?
α-L-iduronidase;
iduronic acid;
heparan sulfate, dermatan sulfate
Sanflippo syndrome types A, B, and C all have to do with improper catabolism of what substrate?
Heparan sulfate
(a glycosaminoglycan/mucopolysaccharide)
Sanfilippo syndrome types A, B, and C all result due to defective catabolism of heparan sulfate hexosamines.
Describe the problem in each type in a stepwise manner.
A - inability to remove a sulfur group
C - an inability to add an acetyl group in place of the removed sulfur group
B - inability to cleave N-acetylglucosamine

Name the enzyme deficient in each of the following:
Sanfilippo syndrome type A
Sanfilippo syndrome type C
Sanfilippo syndrome type B
Heparan N-sulfatase
Acetyl-CoA-acetyl transferase
α-N-acetylglucosaminidase

Hunter and Hurler syndromes both involve removal of the __________ (hexuronic acid OR hexosamine) in the dermatan/heparan sulfate repeating disacharrides.
Sanfilippo syndromes A, B, and C all involve removal of the __________ (hexuronic acid OR hexosamine) in the heparan sulfate repeating disacharrides.
Hexuronic acid;
hexosamine
What is the basic structure of a glycosaminoglycan (mucopolysaccharide)?
(Hexuronic acid + hexosamine)n
(1st position + 2nd position)repeating
Name the enzyme deficient in each of the following:
Sanfilippo syndrome type B
Sanfilippo syndrome type C
Hurler syndrome
Sanfilippo syndrome type A
Hunter syndrome
N-acetylglucosaminidase
Acetyl-CoA-acetyl transferase
α-L-Iduronidase
Heparan N-sulfatase
Iduronate sulfatase
Name the glycosaminoglycan (mucopolysaccharide) that is not being properly catabolized in each of the following:
Sanfilippo syndrome type B
Sanfilippo syndrome type C
Hurler syndrome
Sanfilippo syndrome type A
Hunter syndrome
Heparan sulfate
Heparan sulfate
Heparan sulfate + dermatan sulfate
Heparan sulfate
Heparan sulfate + dermatan sulfate
What glycosaminoglycan (mucopolysaccharide) is not being properly catabolized in Morquio syndrome types A and B?
Keratan sulfate
What keratan sulfate catabolic enzyme is deficient in Morquio syndrome A?
What keratan sulfate catabolic enzyme is deficient in Morquio syndrome B (GM1 gangliosidosis)?
Galactose 6-sulfatase
β-galactosidase
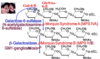
Name each individual disorder (and substrate glycosaminoglycan) for a deficiency of each of the following:
β-galactosidase
α-L-Iduronidase
Acetyl-CoA-acetyl transferase
N-acetylglucosaminidase
Iduronate sulfatase
Galactose-6-sulfatase
Heparan N-sulfatase
Morquio syndrome B (keratan sulfate)
Hurler syndrome (heparan or dermatan sulfate)
Sanfilippo syndrome C (heparan sulfate)
Sanfilppo syndrome B (heparan sulfate)
Hunter syndrome (heparan or dermatan sulfate)
Morquio syndrome A (keratan sulfate)
Sanfilippo syndrome A (heparan sulfate)
Name the enzyme deficient in each of the following mucopolysaccharidoses.
Morquio syndrome B (keratan sulfate)
Hurler syndrome (heparan or dermatan sulfate)
Sanfilippo syndrome C (heparan sulfate)
Sanfilppo syndrome B (heparan sulfate)
Hunter syndrome (heparan or dermatan sulfate)
Morquio syndrome A (keratan sulfate)
Sanfilippo syndrome A (heparan sulfate)
β-galactosidase
α-L-Iduronidase
Acetyl-CoA-acetyl transferase
N-acetylglucosaminidase
Iduronate sulfatase
Galactose-6-sulfatase
Heparan N-sulfatase
Name the enzyme deficient in each of the following mucopolysaccharidoses.
Hurler syndrome (heparan or dermatan sulfate)
Sanfilippo syndrome C (heparan sulfate)
Hunter syndrome (heparan or dermatan sulfate)
Morquio syndrome A (keratan sulfate)
α-L-Iduronidase
Acetyl-CoA-acetyl transferase
Iduronate sulfatase
Galactose-6-sulfatase
Name the enzyme deficient in each of the following mucopolysaccharidoses.
Morquio syndrome B (keratan sulfate)
Sanfilppo syndrome B (heparan sulfate)
Morquio syndrome A (keratan sulfate)
β-galactosidase
N-acetylglucosaminidase
Galactose-6-sulfatase
Name the enzyme deficient in the following mucopolysaccharidosis:
Hunter syndrome (dermatan sulfate and heparan sulfate)
Iduronate sulfatase
Name the enzyme deficient in the following mucopolysaccharidosis:
Hurler syndrome (dermatan sulfate and heparan sulfate)
α-L-Iduronidase
Name the enzyme deficient in the following mucopolysaccharidosis:
Sanfilippo syndrome A (heparan sulfate)
Heparan N-sulfatase
Name the enzyme deficient in the following mucopolysaccharidosis:
Sanfilippo syndrome C (heparan sulfate)
Acetyl-CoA-acetyl transferase
Name the enzyme deficient in the following mucopolysaccharidosis:
Sanfilippo syndrome B (heparan sulfate)
N-acetylglucosaminidase
Name the enzyme deficient in the following mucopolysaccharidosis:
Morquio syndrome A (keratan sulfate)
Galactose 6-sulfatase
Name the enzyme deficient in the following mucopolysaccharidosis:
Morquio syndrome B (keratan sulfate)
β-Galactosidase
Are Hunter and Hurler disease defects in the catabolism of the acidic sugar or the amino sugar of dermatan/heparan sulfate?
Acidic sugar
Are the Sanfilippo (types A, B, and C) disease defects in the catabolism of the acidic sugar or the amino sugar of heparan sulfate?
Amino sugar
Are the Marquio (types A and B) disease defects in the catabolism of the acidic sugar or the amino sugar of keratan sulfate?
Acidic sugar
Name the various drugs used for enzyme replacement therapy for the following conditions:
Hunter syndrome
Hurler syndrome
Morquio syndrome A
Elaprase
Aldurazyme
Vimizim
What is the main component of bacterial cell walls?
Peptidoglycan
Which has a thicker peptidoglycan wall, gram-negative or gram-positive bacteria?
Gram-positive

Describe the difference in structure between gram-positive and gram-negative bacteria.

What is the basic structure of bacterial peptidoglycan?
Repeating disacharrides (similar to a glycosaminoglycan/mucopolysaccharide) cross-linked by peptides
What enzyme creates bacterial walls by cross-linking repeating sacharride units with peptides?
What type of antibiotic inhibits this enzyme?
Transpeptidase;
β-lactams
β-lactamase development provides bacteria resistance against the antibiotic category that targets what enzyme?
What antibiotic category is this?
Transpeptidase (which cross-links peptidoglycan);
β-lactams
What is a ceramide?
What is a glycosphingolipid?
A sphingosine backbone with an attached fatty acid;
a sugar chain linked to a ceramide

What are two subtypes of glycosphingolipid?
Cerebrosides (neutral GSLs)
Gangliosides (acidic GSLs)

Which type of glycosphingolipid contains sialic acid, cerebrosides or gangliosides?
Gangliosides

True/False.
A cerebroside = a ceramide + sugars + sialic acid = an acidic glycosphingolipid
A ganglioside = a ceramide + sugars = a neutral glycosphingolipid
False.
A ganglioside = a ceramide + sugars + sialic acid = an acidic glycosphingolipid
A cerebroside = a ceramide + sugars = a neutral glycosphingolipid
What three sphingolipidoses involve failure to break down cerebrosides (neutral glycosphingolipids)?
Krabbe disease (galactosylceramide)
Gaucher disease (glucosylceramide)
Fabry disease (globotriaosylceramide)
Krabbe disease results as a failure to break down what cerebroside (neutral glycosphingolipid)?
What enzyme is deficient?
Galactosylcerebroside (galactosylceramide);
galactocerebrosidase
Gaucher disease results as a failure to break down what cerebroside (neutral glycosphingolipid)?
What enzyme is deficient?
Glucosylceramide;
glucocerebrosidase
Fabry disease results as a failure to break down what cerebroside (neutral glycosphingolipid)?
What enzyme is deficient?
Globotriaosylceramide;
α-galactosidase A
Type O Tay-Sachs (Sandhoff) disease results as a failure to break down what cerebroside (neutral glycosphingolipid)?
What enzyme is deficient?
Globotetraosylceramide;
β-hexosaminidase (isoform A and B)
Galactose + ceramide =
Glucose + ceramide =
(Glycosphingolipids)
Galactosylceramide
Glucosylceramide

Glucose + ceramide = glucosylceramide
Glucosylceramide + galactose =
(Glycosphingolipids)
Lactosylceramide

Glucose + ceramide = glucosylceramide
Glucosylceramide + galactose = lactosylceramide
Lactosylceramide + galactose =
Lactosylceramide + galactose + GalNAc =
(Glycosphingolipids)
Globotriaosylceramide
Globotetraosylceramide

What is the alternate term for glucosylceramide (a neutral glycosphingolipid)?
What is the alternate term for galactosylceramide (a neutral glycosphingolipid)?
What is the alternate term for lactosylceramide (a neutral glycosphingolipid)?
Glucocerebroside;
galactocerebroside;
lactosylcerebroside
Name the deficient enzyme associated with each of the following cerebrosidoses.
Krabbe disease
Gaucher disease
Fabry disease
Type O Tay-Sachs (Sandhoff) disease
Galactocerbrosidase
Glucocerebrosidase
α-Galactosidase A
β-Hexosaminidase (isoforms A and B)
For each of the following diseases, name the glycosphingolipid that is not properly catabolized:
Krabbe disease
Gaucher disease
Fabry disease
Type O Tay-Sachs (Sandhoff) disease
Galactosylceramide
Glucosylceramide
Globotriaosylceramide
Globotetraosylceramide
The sphingolipidoses involving neutral glycosphingolipids (cerebrosides) basically involve failure to breakdown a compound made of ceramide + sugar monomer(s).
What sugar(s) is(are) involve(d) in Gaucher disease?
(Note: underline any terminal sugars.)
β-Glucose (+ ceramide = glucosylceramide)

The sphingolipidoses involving neutral glycosphingolipids (cerebrosides) basically involve failure to breakdown a compound made of ceramide + sugar monomer(s).
What sugar(s) is(are) involve(d) in Krabbe disease?
(Note: underline any terminal sugars.)
β-Galactose (+ ceramide = galactosylceramide)

The sphingolipidoses involving neutral glycosphingolipids (cerebrosides) basically involve failure to breakdown a compound made of ceramide + sugar monomer(s).
What sugar(s) is(are) involve(d) in Fabry disease?
(Note: underline any terminal sugars.)
α-Galactose + β-galactose + β-glucose (+ ceramide = globotriaosylceramide)

The sphingolipidoses involving neutral glycosphingolipids (cerebrosides) basically involve failure to breakdown a compound made of ceramide + sugar monomer(s).
What sugar(s) is(are) involve(d) in Type O Tay-Sachs (Sandhoff) disease?
(Note: underline any terminal sugars.)
β-GalNAc + α-galactose + β-galactose + β-glucose
(+ ceramide = globotetraosylceramide)

What is the inheritance pattern of Fabry disease?
What is the usual cause of death?
What are some other S/Sy in hemizygous males?
X-linked;
renal and/or cardiac failure;
skin lesions, fever, burning pain in extremities, renal dysfunction
Buildup of what in what tissue type is pathognomonic for Gaucher disease?
Glucocerebroside (glucosylceramide) in tissue macrophages (Gaucher cells)
True/False.
All the neutral glycosphingolipid (cerebroside) sphingolipidoses are characterized by cerebral accumulations of the cerebroside that is not being broken down.
(Note: these diseases include: Gaucher, Krabbe, Fabry, and Type O Tay-Sachs disease)
False.
This is true for each but not Krabbe disease, where a cerebral buildup of galactosylsphingosine (psychosine) is seen instead of galactosylceramide.
Morquio syndrome B (a mucopolysaccharidosis) and Krabbe disease (a sphingolipidosis) both involve deficiencies of a β-galactosidase (specifically, β-galactocerebrosidase in Krabbe disease).
Why do the diseases present differently?
Different genetic causes lead to different phenotypic changes
Krabbe disease –> mutation of the GalCer gene
Morquio syndrome B –> mutation of the GBL1 gene
Type O Tay-Sachs (Sandhoff) disease involves an accumulation of __________________ in what location and/or organelle?
Globotetraosylceramide;
lysosomes
For which of the following cerebroside (neutral glycosphingolipids) sphingolipidoses is there an enzyme replacement therapy available?
Name the medication.
Gaucher disease
Krabbe disease
Fabry disease
Type O Tay-Sachs (Sandhoff) disease
Gaucher d. –> cerazyme
Fabry d. –> fabrazyme
What is the effect of psychosine (galactosphingosine) buildup in cerebral tissues in Krabbe disease (a sphingolipidosis)?
Oligodendroglial toxicity
Mucolipidoses, mucopolysaccharidoses, and sphingolipidoses are all types of _____________ ____________ _____________.
Lysosomal storage disease (LSDs)
How can lactosylceramide (a neutral glycosphingolipid) be changed from a cerebroside to the GM3 ganglioside (an acidic glycosphingolipid)?

By adding N-acetylneuraminic (sialic) acid to the terminal galactose

What are the three major gangliosides (acidic glycosphingolipids)?
GM3 (sialic acid + lactosylceramide)
GM2 (GalNAc + GM3)
GM1 (Galactose + GM2)

What role does GM1 (an acidic glycosphingolipid) play in the body?
It is the major ganglioside of the CNS;
it is a receptor for cholera toxin
Using the attached ganglioside chart (showing GM3, GM2, and GM1), identify the points for which Tay-Sachs and GM1 gangliosidosis occur.

Tay-Sachs: inability to remove β-GalNAc (turning GM2 into GM3)
GM1 gangliosidosis: inability to remove β-Galactose (turning GM1 into GM2)

What is the terminal sugar for GM1 (an acidic glycosphingolipid)?
What is the terminal sugar for GM2 (an acidic glycosphingolipid)?
What is the terminal monomer for GM3 (an acidic glycosphingolipid)?
β-Galactose
β-GalNAc
α-Sialic (N-acetylneuraminic) acid

Which isoform (A or B) of β-hexosaminidase is able to remove β-GalNAc from GM2?
Which requires GM2-activator as a cofactor?

β-Hexosaminidase A;
β-hexosaminidase A
What three enzymes are involved in the various forms of Tay-Sachs disease?
Hexosaminidase A (αβ)
Hexosaminidase B (ββ)
GM2-activator
Here are the two enzymes and one cofactor involved in the various forms of Tay-Sachs disease:
Hexosaminidase A (αβ)
Hexosaminidase B (ββ)
GM2-activator
Which are deficient in each form (Type B, Type O, Type AB)?
Type B (missing α-chains): Hexosaminidase A
Type O (missing β-chains): Hexosaminidase A, Hexosaminidase B
Type AB (neither chain missing): GM2-activator

Here is a description of the missing enzyme (or cofactor*) in each of the three forms of Tay-Sachs disease:
Type B (missing α-chains): Hexosaminidase A
Type O (missing β-chains): Hexosaminidase A, Hexosaminidase B
Type AB (neither chain missing): GM2-activator*
What substrate builds up in each disease, respectively?
Type B (missing α-chains): GM2
Type O (missing β-chains): GM2 + GB4
Type AB (neither chain missing): GM2

Describe the different forms of Tay-Sachs disease (a type of sphingolipidosis).
Normal individual: Hexosaminidase A, hexosaminidase B, and GM2-activator are all functioning normally.
Tay-Sachs Type B (classic Tay-Sachs): α-chain in hexosaminidase A is dysfunctional; the individual cannot digest GM2
Tay-Sachs Type O: β-chain in hexosaminidase A and B are dysfunctional; the individual cannot digest GM2 or GB4
Tay-Sachs Type AB: GM2-activator is dysfunctional; the individual cannot digest GM2

Which type of Tay-Sachs disease is ‘classic Tay-Sachs?’
Which type of Tay-Sachs disease is the most severe form?
Options: Type B (missing α-chains), Type O (missing β-chains), Type AB (missing GM2-activator).
Classic: Type B (missing α-chains) - cannot breakdown GM2
Most severe: Type O (missing β-chains) - cannot breakdown GM2 or GB4

What three blood antigens define the A, B, or O blood types?
A, B, and H
What are the two antigenic possibilities for what is found on the RBC surface of an individual with Type B blood?
- All B antigens
(B/B)
- B and H antigens
(B/H)
What are the two antigenic possibilities for what is found on the RBC surface of an individual with Type A blood?
- All A antigens
(A/A)
- A and H antigens
(A/H)
Individuals with type AB blood can receive straight transfusions of which blood types?
None;
they can receive washed, packed RBCs from any blood type
(the transfusion would still have anti-A or anti-B antibodies depending on the donor blood type)
What is the antigenic possibility for what is found on the RBC surface of an individual with Type O blood?
All H antigens
(H/H)
What is the antigenic expectation for what is found on the RBC surface of an individual with Type AB blood?
A and B antigens
(A/B)
All blood types start with what basic antigen?
How does this antigen get turned into A antigen?
How does this antigen get turned into B antigen?
H (the O antigen)
(Galactose [+ L- fucose] +GlcNAc — RBC);
addition of GalNAc;
addition of galactose

The H antigen has an L-_______ attached to its terminal galactose.
Addition of what sugar to the galactose would result in the type A antigen?
Fucose;
GalNAc

The H antigen has an L-_______ attached to its terminal galactose.
Addition of what sugar to the galactose would result in the type B antigen?
Fucose;
galactose

What occurs in individuals that are unable to add L-fucose to RBC antigens?
The H antigen cannot be created, and, thus, no ABO blood groups result
(Genotype: h/h)
(Bombay disease - antibodies against A, B, and H are in serum)
An inability to add _________ to RBC antigens results in Bombay disease.
Describe Bombay disease.
L-fucose;
an individual has no ABO antigens on their RBC surface and thus has antibodies against A, B, and H
(thus, they cannot receive even type O packed, washed RBCs)

All sphingolipid synthesis start with a simple ___________.
To create sphingomyelin, add ___________.
To create glucosylcerebroside , add ___________.
To create galactosylcerebroside , add ___________.
Ceramide;
phosphocholine,
glucose,
galactose

Sulfatides are a type of sphingolipid made by the ____________ of galactosylcerebrosides in the CNS oligodendrocytes.
If multiple sugars are added to a ceramide, a ______________ results. Adding ________ acid to this results in a ganglioside.
Sulfation;
globoside,
sialic (N-acetylneuraminic)
Type A Niemann-Pick disease results from dysfunction in the metabolism of what substance?
Type B Niemann-Pick disease results from dysfunction in the metabolism of what substance?
Type C1 Niemann-Pick disease results from dysfunction in the metabolism of what substance?
Type C2 Niemann-Pick disease results from dysfunction in the metabolism of what substance?
Sphingomyelin;
sphingomyelin;
cholesterol;
cholesterol
How does type A Niemann-Pick disease manifest?
How does type B Niemann-Pick disease manifest?
More severe form: sphingomyelin accumulation in liver, CNS, and spleen
–> neurodegeneration/intellectual disability and early death;
less severe form: no significant neurological involvement
–> patients typically survive into adulthood
What two enzymes are responsible for sphingomyelin degradation?
Which is deficient in type A Niemann-Pick disease?
Which is partially inactivated in type B Niemann-Pick disease?
Sphingomyelinase, ceramidase;
sphingomyelinase;
sphingomyelinase
What two enzymes are responsible for sphingomyelin degradation?
A deficiency of which enzyme is responsible for Farber disease? What accumulates in this disorder?
Sphingomyelinase, ceramidase;
ceramidase, ceramides
Virtually each sphingolipidosis is due to a defect in metabolic pathways breaking down substrate to what eventual product?
Ceramide

Metachromatic leukodystrophy results from a deficiency of what enzyme?
Resulting in a buildup of what substance?
Arylsulfatase;
sulfatides
