heme/onc Flashcards
(107 cards)
pt presents w -weight loss, night sweats, cough -CXR= apical granulomas w areas of central necrosis surrounded by large cells with abundant pale cyptoplasm most likely dx what are the large cells w pale cytoplasm? what cell marker are they most associated w
TB (apical granulomas = central caseous necrosis surrounded by Møs) macrophages = associated w CD14
pt w sickle cell comes in for fever, dyspnea, chest pain: prior hospitalizations for abd pain were trx w hydration and resolved low hct, high reticulocyte they die in the hospital spleen is found to be firm and brown what is the dx? explain the spleen findings
sx= Acute Chest Syndrome : a vaso-occlusive crises localized to the pulmonary vasculature that can occur in sickle cell patients = commonly precipitated by a previous lung infection -occurs in pts w homozygous HbS = repeated events of vaso-occlusion produce a spleen that is shrunken and fibrotic -spleen is brown bc of hemosiderosis secondary to years of extravascular hemolysis -this is the reason that most homozygous sickle cell patients have undergone “autosplenectomy” by the time they reach adulthood
tissue factor aka ____ plays a significant role in what ds
tissue factor aka thromboplastic associated w DIC: will cause excessive production of thrombin + widespread intravascular fibrin deposition and fibrinolysis –> consumption of clotting factors and platelets
most head and neck carcinomas will met/spread via what method
LYMPH (not direct extension) i.e. tonsils –> jugular LNs
chemo therapy w a cyclophosphomide (a _____-based chemo) has an increased risk of what AE in F patients how will it present how can it be prevented
chemo trx w a nitrogen-mustard based chemo i..e a cyclophosphamide –> HEMORRHAGIC CYSTITIS : inc frequency, dysuria, suprapubic pain, progressive hematuria –> prevent this w aggressive hydration and cotrx w MESNA (2-mercaptoethanesulfonate) that will bind the toxic metabolites of the drug in the urine
in sickle cell anemia, the hemoglobin of cells aggregates in response to what
“sickling” = aggregation of hemoglobin molecules in response to anoxic conditions
- after polymerization, HbS forms a meshwork of fibrous polymers –> RBC distorts into the abn sickle shape
sickling is promoted by…
- low O2 levels
- inc acidity
- low blood volume (dehydration)
(aka the conditions seen in the periphery –> inc sickling in the periphery –> impede BF–> microinfarcts all over the body–> vasoocclusive crises)
-organs that are predisposed to more sickling are
- organs where blood moves more slowly –> low O2 and acidic conditions
- spleen and liver
- organs w high metabolic demand = inc O2 unloading
- brain, M, placenta
compare and contrast the action of unfractioned heparin and LMWH on
- factor Xa
- thrombin
give an example of a LMWH
thrombin = factor I
LMWH= enoxaparin

what is porphyria cutanea tarda
- etiology
- presentation
*
is the MC disorder of porphyrin (heme) synthesis
(porphyria= deficiency in teh early steps of heme synthesis that result in inc of porphyrin-containing metabolites)
- etiology: x uropophyrinogen decarboxylase
- any loss of enzyme after PBG conversion (aka late step dysunction) [see picture] wll result in PHOTOSENSITIVITY bc of the build up of UV LIGHT SENSITIVE PORPHORINS
- presentation:
- acquired > inherited
- risk factors: inc alc intake, smoking, hep C, HIV
- photosensitivity= blisters and vesicle formation on sunexposed areas (i.e. dorsum of hand) : small itchy spots that will blister–> rupture–> heal with hyperpigmentation
- +edema, pruritis, erythema, pain

what is the difference in a heme synthesis defieciency and a porphyria
porphyria: an enzyme deficiency at specific points in the heme synthesis pathway that result in the build up of porphyrins that are UV light sensitive
- late step deficiencies= any enzyme after PBG (PBG deaminase, UP III synthase, UP decarboxylase, CP oxidase
- =photosensitivity= blistering in sunexposed areas + pruritis, hyperpigmentation, erythemia, and pain
heme synthesis deficiency is an xferrochetalase or ALA synthase
- this just presents as a microcytic, hypochromic anemia

child presents w HA and gait ataxia
what is the most likely brain tumor
- classic presentation?
differentiate this from the other common brain tumor in children

pilocytic astrocytomas are the most common brain tumor in children
- HA and cerebellar sx
- imaging shows well demarcated solid and cystic components
- H&E = GFAP+ w hairlike processes and classic rosenthal fibers
VS MEDULLOBLASTOMA
- =the most common malignant tumor in children
- they are exclusively in the cerebellum
- NOT cystic like pilocytic astrocytomas
- imaging will show a solid mass in the cerebellum
- the mass can compress on the v4th ventricle and lead to hydrocephalus (on imaging will look like a huge fluid build up in the superior ventricular system)
- H&E: small cells with hypochromatic nuclei that form homer-wright rosettes

- which drug class will inc PT+PTT but have NO effect on thrombin time
- which drug class will have an effect on thrombin time
- which pathway must be inhibited to effect PT time
- which pathway must be inhbited to effect aPTT time
- direct Xa inhibitors
- direct thrombin inhibitors
- extrinsic
- intrinsic

what are CD3+ cells and what role do they play in the stem cell transplant process
CD3+ = CD4/CD8+ T cells
they are the cells in the donor BM that can cause graft-vs-host ds
(are often depleted from the graft before transplant)
what is the MOA of hydroxyurea
what is its use in sickle cell ds
- hydroxyurea
- has myelosuppressive effects by arresting cell cycle in rapidly dividing hematopoetic cells
- will inc the HbF levels
- effects
- improve O2 delivery
- reduce vasoocclusive crises
- lessen need for transfusions
acute intermittant porphyria
- etiology
- presentation
- trx
- =PBG deaminase deficiency
- hx of abd pain&gen GI sx + neuro sx (change in sleep/concentration, anxiety, numbness+tingling)
- +change in urine (port wine color)
- trx= ALA synthase inhibition
- long term management w glucose or heme will also inhibit ALA

how does HPV associated CA present
what gene mutation does HPV cause
- HPV related CA– sore throat, enlarged/ulcerated tonsil, with a neck mass (often firm and immobile bc malignant)
- HPV is associated w HEAD AND NECK CA
- HPV release E6 and E7 proteins
- E6 protein of HPV degrades p53 from the TP53 gene
- E7== x Rb protein tumor suppressor) from the RB gene
what is the similarity between RET and BRAF
what tumors are they associated with
both are protooncogenes that require GOF mutations to become oncogenes
BRAF
- code fore Ser/Thr specific protein kinase
- melanoma, NHL, papillary thyroid CA
RET
- code for an RTK
- MEN 2A and MEN 2B, papillary thyroid CA
inc 5-HIAA in the urine represents what
the presence of carcinoid tumor
why does warfarin need a bridge therapy w heparin till the INR levels out
having a heparin bridge lowers the risk of what AE
bc warfarin inhibits carboxylaton of NEW k-dependent clotting factors, your body first needs to use up all the pre-existing clotting factors in the plasma
only once those are all gone will the INR reach therapeutic activity
- a bridge will lower the risk of skin necrosis due to the loss of Protein C before anything else (short half life)
compare the change in PT, PTT, platelets, and bleeding time in…
- hemophilia
- vWF deficiency
- DIC
- uremic platelet dysfunction
- heparin use
- warfarin use
- immune thrombocytopenia

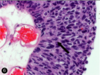
what are the pathologic cells seen in this histo pic
what is the dx?
clinical presentation

spherocytes
- 2/3 the diameter of a normal RBC
- more densely hemoglobinized around the periphery
- no central zone of pallor
- hereditary spherocytosis = x ankyrin and spectrin –> inc sequestration and destruction in the spleen
- inc extravascular hemolysis = jaundice, splenomegaly, inc INDIRECT bilirubin
- often young age
- infection can lead to an acute hemolytic crisis –> acute worsening of pallow, scleral icterus, fatigue, and splenomegaly

describe what an osteoblastic vs osteolytic bone lesion would look like
which metastatic CAs to the bone result in which
osteoblastic = SCLEROTIC = usually more indolent
osteolytic = LUCENT = tend to be from more aggressive CAs

what are the bone changes seen w age
BE SPECIFIC
inc quantity of fat
dec overall mass
desscribe the cause of abn bleeding in patients with uremia
how is this treated
pts w uremia (often ESRD, need dialyses) have abn platelets (qualitatively)
this results in n PT/PTT/platelet count but an abn bleeding time bc platelets are non-functional
treat w dialysis to remove the uremic toxins and allow the partial reversal of the abn
what are the 5 features of anaplastic tumors
- poorly differentiated
- sign variation in size and shape of cells
- abnormally large, hyperchromiatic nuclei
- lots of mitotic figures
- giant, multinucleated tumor cells