Pediatric Neurology Flashcards
Major features of Tuberous Sclerosis
Clinical Triad: Mental Retardation, Epilepsy, Facial Angiofibromas (adenoma sebaceum)
- Adenoma sebaceum seen in 80%
- Periungual fibroma
- Shagreen patch (connective tissue nevus)
- 3 or more hypomelanotic patches
- Multiple retinal nodular hamartomas
- Cortical tubers
- SEGAs (subependymal giant cell astrocytomas)
- Cardiac rhabdomyoma
- Renal angiomyolipoma
- Lymphangiomyomatosis (occurs in females; presents w dyspnea, hemoptysis or spontaneous pneumothorax)
Genetics of Tuberous Sclerosis
Autosomal dominant neurocutaneous syndrome
Two gene loci have been identified for tuberous sclerosis:
- 9q34 (encoding hamartin)
- 16q13.3 (encoding tuberin)
Neurofibromatosis Type I (von Recklinghausen disease)
- NIH criteria: presence of at least 2 of the following—
- 6 or more café au lait spots
- axillary or inguinal freckling
- Optic nerve glioma (pilocytic astrocytoma)
- more than 2 cutaneous neurofibroma or 1 plexiform neurofibroma
- > 2 iris hamartomas (Lisch nodules)
- first degree relative with NF1 by the above criteria
- distinctive osseous lesion—long bone dysplasias, scoliosis, sphenoid wing dysplasia
Minor features: macrocephaly, short stature, learning disability, hypsarrhythmia, epilepsy, scoliosis, hypertension
Risk of neoplasm:
- optic glioma (pilocytic astrocytoma)
- Gliomas are usually astrocytomas of the brainstem, cerebral peduncles, globus pallidus
- ependymoma, meningioma, medulloblastoma
- increased risk of leukemia, Wilm tumor, melanoma, pheochromocytoma, medullary thyroid carcinoma, rhabdomyosarcoma
- risk of malignant transformation of neurofibroma to neurofibrosarcoma is <5% in kids (higher in adults). Presents with rapid growth of lesion & pain
- can see intracranial aneurysms and Moyamoya
Genetics of Neurofibromatosis Type I (von Recklinghausen disease)
- AD, Chromosome 17q 11.2
- mutation on the neurofibromin gene on chromosome 17
Neurofibromatosis Type II
For diagnosis either ONE of the following must be present
1) Bilateral vestibular nerve schwannomas- 90%
2 ) 1st degree relative w NF2 & either of the following in the index case: unilateral vestibular nerve schwannoma; or 2 of the following—glioma, meningioma, neurofibroma, schwannoma
OR TWO of the following must be present
- multiple meningiomas
- unilateral vestib nerve schwannoma
- neurofibroma, glioma, schwannoma, cerebral calcifications or juvenile posterior subcapsular cataracts (schawannomas thus Subcapsular)
(cutaneous findings are less common in NF2 but can see café au lait spots, more commonly see CNS tumors in NF2—schwannoma, meningioma, ependymoma,
Genetics of Neurofibromatosis Type II
AD; chromosome 22
-mutation on merlin gene or schwannomin gene on chromosome 22
Sturge-Weber Syndrome
congenital DO w angiomatosis of face, eye, & meninges
- (considered a congenital malformation rather than a genetic DO and is not hereditary) 2/2 the persistence of embryonal blood vessels that normally regress during gestation)
- ipsi facial port wine stain in distribution of V1 present at birth
- ipsi leptomeningeal angioma (causing contra seizures +/- hemiplegia, homonymous hemianopia)
- progressive venous infarction of the brain underlying the leptomeningeal angioma leads to atrophy & calcification
- ocular malformations may cause glaucoma, optic atrophy, or iris heterochromia
- Glaucoma & ketchup fundus
- Choriodal angiomas
- learning difficulties, developmental delay, dental abn, skeletal lesions
- HA is common; hydrocephalus from increased venous pressure or from extensive AV anastomosis
- MRI w gad: leptomeningeal enhancement, white matter abn, & unilateral hypertrophy of choroid plexus (vascular malformation may be difficult to see)
- Szs in 90%; caused by the leptomeningeal angiomas
- when these angiomas are calcified they appear like “railroad tracks” on CT
Fragile X syndrome
- Most common inherited form of mental retardation
- Expansion of CGG repeats in familial mental retardation 1 gene (FMR1) on chromosome X
- X-linked recessive
- Males 1/4000, Females 1/8000
- females less likely to have impairment and physical features
- males have elongated faces, high forehead, protruberant ears, enlarged testes (BIG EGGS)
Fabry’s Disease
X-linked recessive (lysosomal storage DO)
α galactosidase A deficiency
Think of “SPECKS”—because they have “specks” in theirs swimming trunk area
o S STROKE
o P Pain—painful peripheral neuropathy, GI pain also
o E Eye abnormalities- keratopathy (corneal opacity), cataracts, papilledema
o C Cardiac- cardiomyopathy, valvular disease, arrhythmias
o K Kidneys- ARF, CKD, HTN, uremia
o S Skin- angiokeratomas (tiny painless papules) in swimming trunk distribution—buttocks, thigh, groin, lower abdomen
Tx is enzyme replacement
Genetics of Fabry’s Disease
X-linked recessive (lysosomal storage DO)
α galactosidase A deficiency
Emery-Dreifus Muscular Dystrophy
-X-linked
Defect in gene encoding for EMERIN protein
- contractures at the elbows, ankles, & neck
- cardiac involvement is prominent & have serious conduction abn often requiring a pacemaker
- muscle weakness in upper arms 1st then later pelvic girdle & distal legs
Lennox-Gastaut syndrome
- Age of onset?
- Triad of?
Appears between ages 1 and 10, sometimes de novo, sometimes after infantile spasms.
• Triad of
(1) multiple seizure types refractory to AEDs,
(2) mental retardation (most), and
(3) slow spike-and-wave (1.5-2.5 HZ) activity on EEG.
Landau-Kleffner syndrome
Age of onset?
Clinical Features?
Onset 2-11 years
Language regression - (acquired epileptic aphasia)
- initial word deafness in setting of normal hearing
- develop both receptive & expressive aphasia
Multiple sz types—atypical absence, myoclonic, GTCs, tonic, etc
- EEG: sleep-activated spike discharges. Multifocal cortical spikes, predominantly in a temporal/parietal distribution, frequently bilateral
- Some pts improve with AEDs (Depakote & Lamotrigine)

West Syndrome
triad of infantile spasms, arrest of psychomotor development and hypsarrhythmia

Vitamin B6 deficiency can cause what in infants?
Infantile seizures
can be seen in babies of mothers with Vit B6 deficiency, immediately after the initiation of breast feeding.
- Tx with pyridoxine is therapeutic and no need to start AEDs for baby
- see Vit B6 def in those that are alcoholics, malnourished (lack of fruit, grain veggies) and elderly
What are the 4 skin findings in TS?
Ash Leaf Spots
Adenoma Sebaceum
Shagreen Patch
Ungual Fibromas

Patient has MR, seizures and some abnormal skin findings. She died.
What lesions do you see on her brain?

Cortical Tuber
(Patient had Tuberous Sclerosis)
What condition are these brain findings seen in?
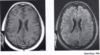
Tuberous sclerosis
subependymal nodules
white matter lesions following lines of neuronal migration, cyst like white matter lesions
