heme portion Flashcards
Iron Deficient Anemia

what size and color? what two unique characteristics about this cell type hint: shape? what are 3 main causes? what should you always suspect? what are 5 unique presentations you might see on PE? what is the treatment? for how long?
hypochromic, microcytic anemia (since no iron to give shape or color)
anisocytosis (unequal size) and piokilocytosis (tear drop shapped)
common causes: blood loss (menses, occult from colon, esophagus, stomach), pregnancy, vegan diet
**always suspect malignancy**
pica (eating dirt/paint), cheilosis, koilonchia “spoon nails”, glottitis “smooth tongue”, esophageal webs, pallor, tachycardia
treatment: Ferrous sulfate 325 mg 3x a day, vitamin C to make absorb better and uptitrate, OR GLUCONATE which is IM or IV
TREAT FOR 6 MONTHS!!

what can cause blood loss in iron deficient anemia? 4 things
GI blood loss from NSAIDS, PUD, cancer
blood donation
trauma
menses
what would you see on the labs for iron deficient anemia?
5 things!
- Low Iron
- High TIBC (since none to bind, very avaliable)
- low ferritin (since none to store)
- low reticulocyte
- hypochromic, microcytic cells
Vitamin B12 deficient anemia

what size and color at these RBC? what factor sets this appart from folate deficient anemia and what are the 5 presentations? what are the 6 things that can cause this and where are the two general sections of the GI system that are effected? what are the 4 important lab results that point to this?
macrocytic/megloblastic anemia, normochromic
can occur from vegan diet, bariatric gastric surgery (MOST COMMON WAY TODAY), ilium resection, chrons disease, pernicious anemia (no intrinsic factor), gastritics
neurologic symptoms, stocking glove paresthesia, loss of position, vibratory sense, balance, glottitis
On lab exams find:
1. antibodies for intrinsic factor
2. MCV >103
2. serum b12 low
4. multinucleated neutrophils!!! 5-6 lobes
Tx: oral supplement or cyanocobalamin nasal spray
what is the number one cause of iron deficient anemia? what are 3 other causes?
- blood loss! need to find the cause!!
2. malignancy! need to think about this
- dietary, vegan!! less common
- poor iron absorption/ trauma
explain how B12 is absorbed and which conditions effect these stages?
B12 is bound to intrinsic factor that prevents it from being absorbed until it reached the ilieum
chrons disease, ilium ressection effect: where it is absorbed
gastric surgery, pernicious anemia, gastritis effect: where intrinsic factor is made
***both of these cause B12 deficiency either in its protection or its absorption!***
what cells produce intrinsic factor that are important for the absorption of B12? where are they located?
parietal cells in the stomach
gastritis PUD can prevent this from working
no intrinsic factor, no absorption
what are three things can cause poor absorption of B12 for anemia?
alcoholism
fish tapeworm
elevated LDH
what are the 3 treatment options for B12 deficient anemia?
- life long vitamin B supplement IM monthly (1000 ug)
2. cyanocobalasmin nasal spray
***neuro symptoms are reversible if treated within 6 months***
explain the schillings test and what it tells you?
tells you which there is a B12 deficiency
Normally, give B12 IM injection so the body is saturday, then give oral B12 radioative, it will be absorbed by the body and AT LEAST 10% excreted in the urine since it isn’t needed. This means uptake is normal and working!!
if pernicious anemia or imparied absorption: LESS than 10% in the urine since not absorbed or partially from bowel
if give intrinsic factor and see B12 in the urine then absorption has improved: pernicious anemia
if no B12 in the uring after intrinsic factor then: problem with absorption in illium, not intrinsic factor
sickle cell anemia
what is the inhertiance pattern? what is the difference between heterozygous/homozygous? when do the problems first occur? where is the mutation? what are 8 things that can prompt sickeling? what are 7 presentations you can see with this disorder?
autosomal recessive
heterozygous 1 Hb S gene: 40%
homozygous 2 Hb SS gene: 80-95%
problems start about 6 months after birth during transition from Hb-F to Hb
mutation in B chain, cause it to sickle under/from:
dehydration, hypoxia, acidosis, infection, temp changes, exertion, alcohol, medical procedures
causes acute painful syndrome, acute chest syndrome, splenic sequestration, aplastic crisis, hemolytic crisis, hand foot disease, “silent” cerebral infarction (35%, subtle but permanent)
what is this and what condition do you commonly see this with?

hand and foot syndrome seen with sickle cell
commonly the first presentation
soft tissue swelling with new bone formation and moth eaten lytic process at proximal aspect of fourth phalanx
no leukocytosis or erythema with the swelling
explain the pathphys of sickle cell
increased RBC destruction
inability to maintain hemoglobin
sickling of cells=increased blood viscosity and ostruction
MORE FRAGIL=hemolysis!
what is the life expectancy for a pt with sickle cell?
40-50 years
die young from infections
Explain actute painful crisis and acute chest syndrome seen in sickle cell pts
acute painful crisis:
excrutiating, can occur anywhere
acute causes vasco occlusion and ishchemia
acute chest syndrome
25% of deaths!
respiratory distress
explain aplastic crisis seen in sickle cell and the five things that can cause it?
stop of RBC production, and since their RBC live so much short ~20 days, they get EXTREME drop in hemoglobin causing aplastic crisis
parvarovirus B19, infection, bone marrow toxins HPV, folic acid deficiency
what do sickle cell patients need to avoid?
altitiudes over 7,000 feet and deep sea diving!
induces sickling
what do you see for lab results for a patient with sickle cell? 6 things!!
- howell jolly bodies
2. Hgb S >50%
3. hb 6-8
4. RBC last 10-20 days
- high reticulocytes
- high ferritin/serum bilirubin
what treatment options are avaliable for someone with sickle cell?
- hydration
- pain meds!!
- transfusion
- folate supplementation
- iron chelation if Fe overload
- preventative vaccines for S. pneumonia, H. influenzae
-prophylatic penicillin from birth to 6 years
hydroxyurea
explain hydroxyurea for sickle cell patients 3 things
- Decrease DNA synthesis
- Inhibit sickling
- Increase Hb F inhibits Hb sickling
**prevents complications and increases life span**
what are sickle cell patients are increased risk for?
(6 things)
- infection with encapsulated organisms
- aseptic necrosis
- CVA
- chronic leg ulcers
- splenic infarc THESE PATIENTS ARE ASPLENIC (DON’T HAVE A SPLEEN THAT WORKS WELL SO NEED TO MAKE SURE THEY ARE VACCINATED ESP AGAINST STREP PNEUMONIA
- retinopathy
explain HbSC and HbSS in sickle cell
Hb SC is the trait for sickle cell, heterozygous and range of symptoms vary
Hb SS the disease for sickle cell, homozygous, severe disease
folate deficiency
what is the most common cause of this? what are four things that can cause this? what size are the cells? what two things do you see on the labs? what 5 drugs can cause this? what is the treatment?
most often caused by poor dietary intake, low fruits and veggies
absorbed in the ileum
macrocytic, hypersegmented PMN, folate
alcoholics, defective absorption (chrons, ulcerative collitis), pregnancy, folic acid antagonist drugs
drugs: methotrexate, alcohol, phenytoin, trimethoprim-sulfamethozole, sulfasalazine
tx: 1 mg folic acid a day
what are lymphomas? where are they?
localized to lymphnodes
malignancy of matured lymphcytes not precursor cells
arises in the PERIPHERY, then can spread to bone marrow
what percent of lymphomas are non hodgkins lymphoma?
90%
burkitt’s lymphoma
(non hodgkins)
where is this an endemic? what type of cell is imporant to see here? what virus does this have a strong association with?

endemic in Africa
“starry sky”, abdominal fullness-cells contain lipid vacuoles!! KEY
EBV association
Type 1: African Jaw/face-100% with EBV associated with malaria
Type 2: sporadic abdomen/bone marrow; 20% EBV
Type 3: HIV associated 40% EBV lymph node/bone marrow

burkitt’s lymphoma
(non hodgkins)
what is the genetic explaination for this and what gene has increased expression?
translocation of chromosome 8 and 14 leading to disregulated c-MYc genes which become protooncogenes and increase expression of Myc
what are two risk factors for non hodgkins lymphoma?
immunosuppression
HIV
hodgkin’s lymphoma
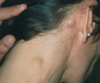
what are four characteristic findings of this? what is this closely associated with? what two tests can you do? what are the two treatment options? what is the age group?
- reed sternburg cells “owl eyed”
- contigious spread, typically cervical, mediastinal, and supraclavicular first effected (moves to neighboring lymphnodes)
- pain after alcohol consumption
- pelbestein fevers (fluctuate over extended periods of time)
EBV association inf 50%
bimodal age group, peaks in 20s then again in 50s
DX: Ct abdomen/pelvix/chest or PET CT, BM biopsy
TX: radiation or chemo depending on the stage (described later)

explain the ANN arbor staging for lymphoma? how does this impact the treatment for hodgkins lymphoma?
if the staging for hodgkins lymphoma is 2a or above RADIATION!!!
2b and worse=ABVD CHEMO/ BEACOPP chemo!!!
(just FYI, this isn’t effected in non-hogkins lymphoma)

mutiple myeloma
what is the pnuemonic associated with this cancer and what does each part mean? what cell is affected here? what are two important test results you will see? what are the two treatment options for this? what group of people is this most common in?
BREAK ACRONYM-malignany of plasma cell, more common in african american men
B: bone pain-lytic bone lesions, increased bone fractures particullary in back, spine, ribs
R: reccurent infections strep pneumoniae, gram neg encapsulated, non function Ig
E: elevated calcium since bone being destroyed
A: anemia (crowding out of bone marrow, overgrows RBC/platelets=less RBC)
K: kidney failure (increased Ig deposit in kidney, increased viscosity=kidney failure)
monoclonal Ig Spike, “M” protein spike, bence jones proteins in the urine,
TX:chemo, BM transplant (not commonly done since many patients elderly)

Acute lymphacitic leukemia (ALL)
what is this the most common of? what subsets of cells does it involve? what will you see on the smear? what condition is it associated with? what is one caution you NEED to address when treating this? what is used for diagnosis and what is the treatment?
1 cause of cancer in children!!
very young: 3-7 yrs
associated with down syndrome
T or B lymphblasts subsets
**hides in CNS so must treat with prophylaxis!
bone pain since ramped up
DX: smear, BM biopsy
TX: 3 phases of chemo!! prophylaxis chemo Ara-c for cancer hiding in CNS, LUMBAR PUNCTURE FOR CNS INVOLVEMENT!!!

explain the two T and B cell subsets of acute lymphocytic leukemia!!
what are the chromosomes that are effected in B cell ALL? what are the symptoms associated with T cell ALL?
This was from khan academy and meded

Acute myelogenous leukemia (AML)
who do you see this in? what are two important things you will see on the SMEAR? what percent achieve remission? what are the three treatment options? what exposure do you usually have to get this?
adults, acute picture
>20% blasts, AUER ROD CELLS, bone pain since ramping up cells
WBC>100,00
70% achieve remission
EXPOSURE: BENZO, CHEMO, RADIATION
TX: combination chemo, bone marrow transplant vit A

chronic lymphocytic leukemia (CLL)
who is this most common in? what type of cells are most common? what is important cell seen on slide? what are three other presentations of this? what do you do for treament of this for the 3 different pt groups? what do the symptoms come from? what is a halmark here?
elderly, asymptomic, die with it not from (3-10 years LE)
95% B cell derived!
B cell “smudge cells” (not structually strong since immature)
hypgammaglobinemia (don’t make as many since B cells messed up)
affects the bone marrow, liver, spleen, lymphnodes! gets bigger
in the lymph node starts a small lymphocytic lymphoma the transfors by richter transformation to diffuse B cell lymphoma (SOLID MASS)
causes autoimmune hemolytic anemia (nonfunctioing antibodies attack RBC, random, but imporant!)
acceptable to wait and watch in asymptomatic
usually come to the office for something else and are diagnosed on accidental findings
symptoms come from overcrowding->thrombocytopenia/anemia->beeding
>65 asymptomic: wait and watch
>65 symptomatic: chem
TX: chlorambucil/fludarabine if chemo

chronic myelogenous leukemia (CML)
what two genes are associated wit this and which one is more specific? which line of cells can this be in and which is the most common? what are the three stages for this? what are the two treatment options? what kills these patients?
Adults
- philidelphia chromosome (95) 9/22
- BCR-ABL gene
- relies on tyrosine kinase inhibitor
- most common in neutrophils, but can be in ANY of the myeloid cell lines
- blast crisis which transforms it to acute myelogenous leukemia **this is what kills these patients**
WBC>100,000
Three stages:
- chronic: asymptomatic
- accelerated: start to see symtoms
- acute: blast crisis! >30% blast in the BM, then transforms to AML
TX: TYROSINE KINASE INHIBITOR IMATNIB (works on BCR-ABL), ALLOGENIC BONE MARROW OR STEM CELL TRANSPLANT

sideroblastic anemia (lead poisoning)

what are the size of the cells? reticulocyte count? platelets? what are three unique lab results you will see with lab testing? what will the patient present with for symptoms? what do you do for treatment depending on the levels of lead in the blood? what does the location of the lead tell you about how long it has been in the body? what does the bone marrow produce?
microcytic, decreased reticulocytes, low platelets
basophilic stippling, elevated lead, erythrocyte protoporphyrin, bone produces ringed sideroblasts instead of health RBC
PE: lead on the gum lines, vomiting, abdominal pain
high serum levels: acute attack
if in the bone: hard to tell how long its been there
BLL>20, medical and environmental intervention
BLL>45, chelation

G6PD deficient anemia
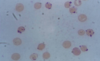
what genetic link is this connected with? what two populations of people is this common in? what size are the cells? what is one unique presentation do you see? exposure to what 3 things can cause this? what are the 5 things you will see on the lab results? how is it treated?
x-linked recessive, african america males (12%), or greeks/mediteranean (20-30%)
normocytic anemia, increased risk of HEMOLYSIS UNDER STRESS,jaundice
G6PD protects RBC agains oxidative stress that can damage the RBC beyond repair
fava beans
infection
oxidative drugs
heinz bodies, low G6PD, bite cells, increased reticulocytes and serum bilirubin
Tx: self limiting, when the the stressor is resolved then normal RBC produced again

beta thalaseemia

what chain is effected in this? what parts of the world is this common in? what unique cell do you see in the lab results? what are the two main classifications of B thalaseemia? when is the more severe on diagnosed? what are four clincial presentations of this? what are 3 treatment options? what do you want to keep [hb] at? what test do you use to tell the difference between Fe and thallessemia?
deficient synthesis of B-globin chain of hemeoglobin
(results in increase A) African/mediteranneans
HEINZ BODY CELLS!! “Target cells” “HAIR ON END APPEARANCE ON XRAY”, FRONTAL BOSSING
minor: heterozygous, sufficient Hb sythesis
major: homozygous! SEVERE transfusion dependent anemia, diagnosed 1st year of life when hb F turns to Hb A (adult) “cooleys anemia”
growth retardation, hepatosplenomegaly, abnormal facial formation, fractures/osteopenia, delayed or absent puberty, hypogonadism
Tx: regular blood transfusions to keep hemoglobin at 12 mg/dl, avoid Fe supplements, bone marrow transplant/splenectomy
MENTZER INDEX

xalpha thalessemia

what are the deficient in? what type of cells can be present? what nationality of people are most common? explain the four stages? size and color? what lab results remain normal? what do you treat with? what should you avoid?
deficient a-globin chain, 4 stages target cells Heinz bodies!!
ASIANS
usually diagnosed if iron supplemets for suspected iron deficient don’t work
- silent carrier, 1 gene deleted
- trait, 2 gene deleted leading to mild hemolytic anemia
- Hb “H”, 3 genes deleted, hemolytic anemia without transfusion need
- lethal at birth, hydrops (seen in pic)
microcytic hypchromic but not very anemia, normal iron, TIBC, ferritin
Tx: folic acid supplement, avoid iron, if that doesn’t work then transfusion but not dependent like B thalassemia

