Gunn: Clinical Endocrinology Flashcards
What is insulin
Insulin is a hormone made by the pancreas that allows your body to use sugar (glucose) from carbohydrates in the food that you eat for energy or to store glucose for future use. Insulin helps keeps your blood sugar level from getting too high (hyperglycemia) or too low (hypoglycemia).
Describe the pathology of Type 1 Diabetes Mellitus
Type 1 diabetes is a chronic illness characterized by the body’s inability to produce insulin due to the autoimmune destruction of the beta cells in the pancreas. Onset most often occurs in childhood, but the disease can also develop in adults in their late 30s and early 40s.
- Infiltration on WBC can be seen over time where the B-cells get progressively destroyed

What is the time course of T1DM and how does it vary for adults vs children
- Young kids have over 90% loss of B-cells
- Slower onset in older patients: and less of a loss (80-90%)
- Progressive loss of B-cells
- Honeymoon Phase - 90% of cells have been destroyed, but 10% is still active = we are getting exogenous insulin but the body is still doing some self-functioning
- Upregulation of T cells
- Increase in related antibodies

Glucose and lipid metabolism are regulated by ______ and it’s counter-regulatory hormones :
Glucose and lipid metabolism are regulated by Pancreatic hormone insulin and it’s counter-regulatory hormones :
- Glucagon
- Catecholamines
- Cortisol
- Growth hormone
Insulins action?
stimulates:
- Glucose uptake in muscle/adipose tissue
- Glycolysis
- Glycogen synthesis
- Protein synthesis
- Uptake of ions (esp K+ and PO4-3)
- Uptake of amino-acids for growth and development of muscle
Inhibits:
- Gluconeogenesis: generation of glucose
- Glycogenolysis: breakdown of glycogen to glucose-6-phosphate and glycogen
- Lipolysis
- Ketogenesis
- Proteolysis
Highly anabolic, lots of athletes stupidly try and use, which induces hyper glycemia

Describe Glucose and insulin level differences over 24 hours
- Glucose/ blood sugar are not stable throughout the day; and they are always dynamically changing
- Increase after breakfast etc
- Fasting vs fed levels are quite different
- Fluctuations of blood sugar and insulin complement each other. We are unable to manage the fluctuations and we don’t want to, as they create important gradients
- Your lowest blood sugar = pre-breakfast blood sugar (and then tends to be higher over the day)

How does glucose and insulin get in cells?
Glucose: In via Glucose Transporter-4 (GLUT-4)
Insulin: in via Insulin receptor
- Our peripheral tissues are essentially impermeable to glucose = stops glucose being taken up
- When sugar goes up, insulin is released, it ‘unlocks/inserts’ GLUT-4 and glucose can diffuse passively down the gradient into cell
- Also stimulates other things, lots of other effects of insulin (see the other slide)

How do we make ketones? an overview
Ketones; (acetoacetate, beta-hydroxybutyrate, and their spontaneous breakdown product, acetone)
- We all make a small amount of ketone as a glucose sparing fuel (to preserve it for brain, although the brain can handle some ketones)
- Before birth, 20% of cardiac energy comes from metabolites and ketones
-
Lipolysis of “white fat” (mass of triglycerides) - Hormone-sensitive lipase is turned off by insulin which turns off lipolysis, activated by glucagon, GH, glucacorticoids.
- Growth Hormone is a potent lipolytic hormone, hence its abuse by bodybuilding
- lack of inhibiting insulin leaves lipase available to be stimulated by any small amount of glucagon or other stimulator
- Fatty acids go to the liver; if you have a lack of insulin + counterregulatory hormones + an excess of glucagon, they will go through B-oxidation.
FA breakdown → acetyl-CoA. The 3 Ketone bodies are produced from acetyl-CoA, mainly in the mitochondrial matrix of liver cells, in excess when carbohydrates are so scarce that energy must be obtained from breaking down of fatty acids.
making ketones in FAT
- ‘Mobilise’ fat
- Stimulate B-oxidation (glucagon)
- Due to the lipolytic effect of growth hormone, its administration in man has been reported to increase plasma nonesterified fatty acid (NEFA) concentrations. Ketone body production increases during acute growth hormone excess as a result of increased NEFA concentrations;

Making ketones in the LIVER:
- Done via an increase in FA oxidation
Ketones require:
- Lack of insulin AND excess glucagon

Making ketones in the mitochondria:
FFA → acetyl CoA
- Three ketones (in green)
- Acetoacetate and B-hydroxybutyrate are acids, acetone is not
- Acetone = what’s usually smelt on breath ( paint smell), which is genetically controlled (about 10% of people cannot smell acetone or paint)
- Acidosis accelerates acetoacetate → B-hydroxybutyrate

What is the anion gap in ketone production
The anion gap is affected by changes in unmeasured ions. A high anion gap indicates acidosis. In uncontrolled diabetes, there is an increase in ketoacids due to the metabolism of ketones. Ketoacids are unmeasured anions, so there is a resulting increase in the anion gap.
- B-hydroxybutyric acid and acetoacetic acid are acids and therefore dissociate completely. (They are in a dynamic balance, with more H+ combining with NADPH to make NAD and pushes acetoacetate to B-Hydroxybuturate)
- Excess H+ + HCO3- → CO2 + H2O = decreased [HCO3-]
- Ketone bodies circulate as anions (as the H+ has been taken away) → increased anion gap
- Formula Anion Gap = Na+ - (Cl- + HCO3-)
- Normal anion gap is 12 (+/- 2) mmol/L: AG tells us when there’s unmeasurable anions are floating around
- THUS in DKA: bicarbonate is replaced by B-hydroxybutyric acid and acetoacetic acid.
- Decreased HCO3- approximates increased anion gap

What happens to the Chloride?
- If B-hydroxybutyric acid dissociated and H+ is converted to CO2 and H2O
What is left: Na/B-hydroxy
- Na-β-hydroxybutyrate is lost in urine…..
- Hydroxybuturate (acid) binds with Na to make a salt = the salt is circulating, not the acid. Some stays in the blood, the rest is filtered by kidney and excreted in urine
- What happens to Chloride?
- In short-term the Cl remains in ECF , therefore no change. DKA patients initially have hyperchloremia. Na is lost in urine, but we normally infuse +++
- 0.9% NaCl, i.e. in excess it replaces Na → hyperchloremia (short term) is always observed in diabetic patients post-treatment
-
Salt loading means hyperchloremia is common after DKA
- Not considered dangerous or associated with mortality
Urine “ketones” = Acetoacetate
- “Ketone” strips use nitroprusside reaction to produce a purple color in the presence of acetoacetate (AcAc), not even measuring all ketones. Normally this is fine, but if you’re acidotic your AcAc is pushed towards B-hydroxy and so the amount of ketones becomes grossly underestimated
- Acetone is detected only if the reagent contains glycine in addition to sodium nitroprusside; doesn’t detect ß-hydroxybutyrate (BOHB)
- Normally, serum BOHB: AcAc ~ 1:1
- Acidosis BOHB: AcAc 1.3:1 to 5.5:1
- Plasma or urine AcAc concentration alone underestimate the severity of ketonemia. So now we measure B-hydroxybuturate alone
Now we can measure B -hydroxybutyrate directly, what do we see when we add insulin!!
- Typical levels of BOHB during DKA
- Steady fall after treatment with insulin, reaching 3mmol/L after ~8/9 hours, then gradually returning to normal levels by 24h
- Bedside tool underestimated the initial level - quite accurate at the level where you’re meant to do it (when BOHB is <3mmol/L), but terrible when looking at severe acidosis. Designed for the lower levels.
- Is this patient developing ketones? You can speed this test up by 2 hours by adding somatostatin and blocking glucagon. But it isn’t really needed when we already have the insulin

Criteria to diagnose DKA
- Current:
- hyperglycemia (blood glucose>11mmol/L)
- Venous pH <7.3 or bicarbonate <15mmol/L
- The presence of ketonemia or ketonuria
- Suggested:
- hyperglycemia (blood glucose >11mmol/L)
- venous pH <7.3
- Serum BOHB >3mmol/L
Considering how useless urine screening is at detecting severe DKA, and that bedside measurers are available everywhere, it’s suggested we should replace “presence of ketonemia” with “serum BOHB breath screening>3mmol/L”
DKA= relative lack of insulin + stress
NOT JUST LACK OF INSULIN BY ITSELF
Insulin (anabolic)
- glucose used for energy substrate or stored as glycogen
- protein formation
- Fats stored as triglycerides
Counterregulatory hormones (catabolic) activated by stress!
- glycogenolysis
- Proteolysis-gluconeogenesis
- Lypolysis: FFA and ketone bodies
You just need a lack of insulin, don’t need to have hyperglycemia
So, if DKA is bad, why is this girl alive?
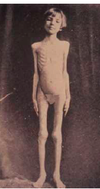
Because she has no FAT. She has had this for 6 months.
DKA requires
- LACK of insulin
- STRESS: To activate counter-regulatory system
- FAT : If no TGs, no NEFA, no Ketone bodies can be made
- This girl has insulin lack but no fat
- She is being starved to death, as a part of her treatment so that she didn’t make ketones
Where is most of the K+ in our bodies?
- In our cells! ICF= 140
- (only 2% ECF, around total daily intake)
- We respond to changes in K+ in order to keep it tightly regulated
- Immediate: shift out of cells
- Late: excrete out of body
- LEARN DIAGRAM

How is the ECF K+ tightly regulated?
- Mean K+= 4.2 mmol/L (3.5-5mmol/L)
- The extracellular pool: 14L
- say K+= 4.2
- 4.2 x 14L = 59 mmol/L (s[K] )
- One meal of fruit /meat ~40-70mmol
- 70mmol/14L ECF = 5mM!!!
- S[K] 4.2 to 9.2 mmol/L a very dangerous rise!
We rely mainly on the feedforward control of K
In Feedback: K in serum increases (increases ECF) → high volume in ECF to try to dilute K to a norm. Excretion won’t occur unless the level in the serum is dangerously high, and we don’t want it to get to this point, feedforward system bypasses the ECF
Feedback vs feedforward
Feedforward keeps K steady by anticipating an influx of K+ (gut senses incoming K). This enables bypassing of ECF to trigger excretion of K; rapid, anticipatory, keeps K stable
** cells can act as a temporary buffer when serum K is too high, they can have a temporary influx of K uptake

What controls K after a meal
-
Insulin → stimulates Na/K ATPase → K uptake
- Not linked to glucose uptake (GLUT4) - just give glucose to stop hypoglycaemia
- Consumption of a meal containing glucose or amino acids stimulates K shift, independent of [K]
- Some evidence for Glucagon & cAMP
- Protein-rich meal→ inc [glucagon] + cAMP
- Glucagon portal vein infusion→ inc transtubular K+ gradient →GFR, → 2 x K excretion
- ? Gut factor?
What is a feedback response to increased K+
Allows a longer term excretion and is needed chronically.
Aldosterone → K uptake by cells (as well as excretion, possible through Na/K ATPase
renal excretion
Other controls for K gradient
-
B2 adrenergic stimulation (Adrenaline)
- shift from ECF → ICF (decr. s[K] )
- exercise can increase adrenaline promoting reuptake into muscles
- Beta blockers tend to increase s[K], prevents reuptake into muscle
- acidosis increases K loss from cells
- Likely by inhibiting Na/K ATPase**
- Cell lysis! eg; muscle, red cells
- K + ICF → ECF
**H/K exchanger does not seem to apply to organic acidosis = likely reason is that as they are organic, they cross the cell membrane and BHOB/acid is both outside and inside cell, hence the exchanger never seems to come into play
What other shifts can increase s[K] ??
- Exercise!
- Physiological: contraction release of K
- Combined with B-blocker or low insulin?
- There is repeated muscle contraction, which leads to K release, repeated massive depolarisation
- Increased ECF osmolarity
- inc ECF osmolarity → water out of cells → increases ICF [K] → increases the gradient driving K+ out of the cell → ECF [K]
- same for all other ions!!
- The reverse is also correct when you’re diluted
Kidneys reabsorption of K

- Most of the reabsorption >65% occurs in the PCT
- Loop of Henle: 25-30% reabsorped
- DCT 4% absorbed, whatever is left is excreted, we excrete~12% due to our diet intake of K is more then we need
**most variation in K excretion happens in the DCT and CD, (but overall is still relatively constant)

Is Renal excretion or retention of K+ faster?
Excretion is faster!
-
The kidneys are able to excrete very large amount of K+
- Maximum secretion can be > GFR filtration
- The Kidney can be slow to conserve K
- Fractional K excretion can be reduced to ~2% of the filtered load (cannot drop it to 0)
- Thus K depletion with hypokalemia can result if K intake is restricted, even with normal people.
How does aldosterone act on the principal cells of the CD, what 3 factors does it affect
- Stimulates activity of the Na/K ATPase on basolateral membrane.
- Increases Permeability of luminal membrane
- Increases electrochemical gradient from lumen → blood

Intercalated cells and K absorption?
- around 10% of epithelial cells in DCT?CD
- Reabsorb K from lumen eg; during hypokalaemia
- Mediated by H+ K+ ATPase???
What controls distal secretion of K+???
- increased s[K+]
- increased distal tubal flow rate]
- Increased aldosterone
Note: acidosis decreases K secretion
and alkalosis increases secretion but is clinically uncommon
How does increased s [K+] → increased K excretion
- Constant physiological correction as K goes up, Aldosterone goes up
- K has a direct stimulation of Na/K ATPase
- Higher gradient, (less back leak)
- Stimulates Aldosterone

How does increased distal flow → increased K secretion?
- Increased flow → decreased K in the tubular fluid
- ie; increased gradient between lumen and blood
- Decreased flow → increased sK in severe renal impairment
- Diuretics: increased flow leads to decreased sK

