Genetics - Musculoskeletal Block Flashcards
What is the simplified pathophysiology of tuberous sclerosis complex?
Mutation in genes coding for tumor suppressor proteins
–> potential mix of benign tumor growth in several organs
(e.g. kidneys, heart, liver, eyes, lungs, and skin)
Does tuberous sclerosis typically result in benign or malignant tumors?
Benign
What are some of the organs most commonly affected by tuberous sclerosis?
Skin, brain, kidney, heart, lung
85% of individuals with tuberous sclerosis will show a mutation in one of what two genes?
TSC2 (69%);
TSC1 (31%)
Name three skin lesions commonly associated with tuberous sclerosis.
Hypomelanotic (ash leaf) macules (87-100% of cases)
Facial angiofibromas (47-90% of cases)
Shagreen (coast of Maine) patches (20-80% of cases)
Name two brain lesions commonly associated with tuberous sclerosis.
Name two signs or symptoms of brain lesions commonly associated with tuberous sclerosis.
Subependymal nodules (SENs; 90% of cases)
Cortical tubers (70% of cases);
Seizures (80% of cases)
Intellectual Disability (50% of cases)
Tuberous sclerosis is associated with mutations in what two genes (85% of cases involving one of these genes)?
What two proteins are coded for by these two genes, respectively?
TSC1 - hamartin;
TSC2 - tuberin
What is the leading cause of increased morbidity and mortality in tuberous sclerosis?
What is the 2nd leading cause of increased morbidity and mortality in tuberous sclerosis?
CNS tumors / intellectual disability;
renal disease
What is the most common renal lesion in tuberous sclerosis?
What is the 2nd most common renal lesion in tuberous sclerosis?
Benign angiomyolipomas (70% of cases)
epithelial cysts (30% of cases)
True/False.
Tuberous sclerosis complex has a fairly high association with autism spectrum disorder?
True (~0.4)
What mode of inheritance does tuberous sclerosis show?
Autosomal dominant
(with variable expressivity)
What is the purpose of the proteins hamartin and tuberin?
What disease is associated with mutations in the genes coding for these proteins?
What are the genes?
Tumor suppression;
tuberous sclerosis;
TSC1 (hamartin) and TSC2 (tuberin)
The following list displays some of the major features of tuberous sclerosis that are present in the majority of cases (bolded).
Below are a few features that aren’t necessarily present in the majority of cases (italicized).
- Subependymal nodule (SEN) (90%)
- Hypomelanotic (ash leaf) macule (87-100%)
- Facial angiofibroma (47-90%)
- Cortical tuber (70%)
- Shagreen (coast of Maine) patch (connective tissue nevus) (20-80%)
- Cardiac rhabdomyoma (47-67%)
- Renal angiomyolipoma (typically benign)
- Multiple retinal hamartoma
- Lymphangiomyomatosi (LAM) (30%)
- Subependymal Giant Cell Astrocytoma (SEGA) (6-14%)
What is a defining feature of tuberous sclerosis autosomal dominant inheritance?
Variable expressivity
What mode of inheritance does tuberous sclerosis display?
What may be responsible for its variable expressivity?
Autosomal dominant;
the ‘two-hit’ hypothesis / random nature of second hit (TSC1, TSC2 genes)
True/False.
The majority of cases of tuberous sclerorsis develop malignant renal angiomyolipomas.
False (<3%);
70% show benign renal angiomyolipomas
Define the medical term proband.
An individual who presents with a genetic disorder
(must warrant investigation of the individual’s family)
What percentage of tuberous sclerosis probands (new cases) have an affected parent?
What percentage do not?
1/3;
2/3 (de novo mutations)
An individual with tuberous sclerosis complex has what likelihood of passing it on to their progeny?
(Note: meaning the likelihood of a single birth where one parent is affected resulting in a child with the disease)
50%
(autosomal dominant)
A child is born with tuberous sclerosis. Neither of her parents are affected. What risk do her future siblings have?
1-2%
(possible germline mosaicism)
What dermatological lesion is here shown?
With what neurocutaneous disorder is it associated?

A shagreen patch;
tuberous sclerosis complex

What dermatological lesions are here shown?
With what neurocutaneous disorder is it associated?

Hypomelanotic macules;
tuberous sclerosis complex

What dermatological lesions are here shown?
With what neurocutaneous disorder is it associated?
Similar wart-like projects and bumps are sometimes associated with this disorder and found where on the body?

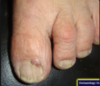
Facial angiofibromas;
tuberous sclerosis complex;
skin around nail folds (periungal fibromas)
A seven-year-old male is brought in by his concerned parents for your evaluation after experiencing a short seizure of undetermined type and origin.
Upon physical exam, you notice the child has several near-white macules on his hips, back, and shoulders. You also notice some roughened skin on his face in a malar distribution.
The child is mentally disabled.
What syndrome is at the top of your differential?
It is associated with mutations in what gene(s)?
Tuberous sclerosis;
TSC1 (hamartin protein), TSC2 (tuberin protein)
What is the treatment for a patient with tuberous sclerosis that has a subependymal giant cell astrocytoma?
What is the treatment for a patient with tuberous sclerosis that has a renal angiomyolipoma?
What is the treatment for a patient with tuberous sclerosis that is experiencing somewhat frequent seizures?
mTOR (mammalian target of rapamycin) inhibitors;
renal artery embolization / surgery (renal sparing);
normal epileptical / seizure control
What disorder has a similar pathophysiology as tuberous sclerosis complex (mutations in tumor suppressor genes/proteins) but is mainly characterized by benign tumors of the nervous system?
Neurofibromatosis
What are the two types of neurofibromatosis called?
Some sources include what as a third type?
Neurofibromatosis type I and type II;
Schwannomatosis
Neurofibromatosis type I is associated with what tumor suppressor gene on what chromosome? What is its inheritance pattern?
Neurofibromatosis type II is associated with what tumor suppressor gene on what chromosome? What is its inheritance pattern?
NF-1, chromosome 17, autosomal dominant;
NF-2, chromosome 22, autosomal dominant
Describe the inheritance pattern of both types of neurofibromatosis.
(E.g. inheritance type, expressivity, penetrance, mutation rate)
Autosomal dominant;
nearly 100% penetrant;
high level of variance in expressivity;
high mutation rate (50% of cases are new cases)
What is the hallmark feature of neurofibromatosis type I?
What is the hallmark feature of neurofibromatosis type II?
Cafe au lait spoits;
acoustic neuromas
What is the hallmark feature of neurofibromatosis type I?
Cafe au lait spots
What is the hallmark feature of neurofibromatosis type II?
Acoustic neuromas
Why is the diagnosis of neurofibromatosis so difficult?
High variance in expressivity
Dianosis of neurofibromatosis type I is made if a person has ____ of the following signs / symptoms:
≥ 6 café au lait spots (> 5 mm in diameter in pre-pubertal persons and > 15 mm in post-pubertals)
≥ 2 neurofibromas of any type
Freckling in the axillary or inguinal regions
Optic glioma
≥ 2 Lisch nodules
A distinct osseous lesion
A first degree relative with NF-1 (by the above criteria)
≥ 2
What is the most common CNS tumor of neurofibromatosis type I?
Optic gliomas
Chromosome 17 produces a protein that is involved in maintaining the balance between cell proliferation and differentiation.
What protein is it and a deficiency in it is associated what disease?
Neurofibromin;
neurofibromatosis type I
Diagnosis of neurofibromatosis type II requires 2 or more of what clinical features?
Either
Bilateral auditory canal masses;
OR
an affected first-degree relative and a unilateral auditory mass
What dermatological lesions are here shown?
This is indicative of what disorder?
It is associated with a mutation of what chromosome?

Cafe au Lait (also known as coast of Maine) spots;
neurofibromatosis type I;
chromosome 17

What is the pathophysiology of Huntington’s disease?
Neuronal death due to Huntingtin protein amyloidosis
What is the genetic cause of Huntington disease pathology?
CAG trinucleotide repeats on chromosome 4
Less than how many CAG repeats on chromosome 4 is normal?
Greater than how many CAG repeats on chromosome 4 is associated with severe Huntington’s disease?
< 26;
≥ 40
What is the inheritance pattern of Huntington’s disease?
It usually presents at what age?
Autosomal dominant’
30 - 40 (can be as low as 10 and as high as 60)
The increased severity of Huntington’s disease is associated with transmission from which parent?
Paternal transmission
Name the major signs and symptoms of Huntington’s disease.
Progressive chorea, rigidity, psychiatric symptoms, and dementia
What is the most common form of inherited cognitive impairment?
Fragile X syndrome
(spectrum of ADHD vs. mild impairment vs. autism vs. severe intellectual disability)
What disorder is associated with trinucleotide CGG repeats on the X chromosome?
What number of repeats is considered normal?
What number is considered pathological?
Fragile X syndrome;
5 - 44;
> 200
Fragile X syndrome is generally more severe in which sex?
Males
Some cells have 100s of _________.
Highly active cells can have 1000s of _________.
Mitochondria;
mitochondria
What organelle is the major source of reactive oxygen species (ROSs)?
What organelle regulates apoptosis?
Mitochondria;
mitochondria
What is homoplasmy?
What is heteroplasmy?
A cell contains only one type of mtDNA (DNAmitochondrial);
a cell contains both normal and mutant mtDNA
What is difficult about the pleiotropic nature of many disorders of the mitochondrial genome?
Different organs can be affected in different patients with the same mutation
Disorders of the mitochondrial genome are often pleitotropic and show variable ___________, making them difficult to both diagnose and also track through generations.
Expressivity
True/False.
A mutant gene must show effects on multiple phenotypic characteristics simultaneously in order to be considered an example of pleiotropy.
False.
(the same pleiotropic gene can show different effects in differing individuals)
If a woman is homoplasmic for a certain mutation, how many of her children are likely to receive that mutation?
If a man is homoplasmic for a certain mutation, how many of his children are likely to receive that mutation?
All of them;
none of them
A woman who is heteroplasmic for a point mutation or duplication will pass it on to how many of her children?
(What will the effect be like?)
All of them
(however, there will be varying severity of phenotypic manifestation as the fraction of mutant mitochondria in the offspring may vary significantly)
A woman who is heteroplasmic for a deletion will pass it on to how many of her children?
(What will the effect be like?)
Likely none of them
(Heteroplasmic deletions are not typically inherited)
What are a few factors that contribute to the wide variability in expressivity in disorders of the mitochondrial genome?
Pleiotropy;
heteroplasmy
(fraction of mutant mtDNA among varying individuals and even varying tissues means there is often a whole spectrum of disease manifestation)
True/False.
The fraction of mutant mtDNA among differing individuals and even differing tissues varies widely, meaning there is often a whole spectrum of disease manifestation.
Yes;
this explains part of the varying expressivity and pleiotropy of disorders of the mitochondrial genome
What are two examples of mitochondrial diseases that result from mutations in nuclear DNA?
(1 category and 1 disorder)
Disorders relating to the POLG gene (involved in mtDNA replication);
Leigh syndrome (75% of cases;
25% of cases come from deletions of mtDNA)
Mutations in the POLG gene often have what effect?
A significant drop in mtDNA in affected tissues
Describe the clinical manifestations of mitochondrial disorders regarding the following terms:
- Inheritance
- Symptoms
- Organ
- Time
- Scenario
- Diagnostics
- Progression
All extremely variable
(due in part to pleiotropy, heteroplasmy, and variable expressivity)
An individual has a significantly low number of mitochondria in each of her cells.
Mutations in what gene may be responsible?
Is there a typical phenotype for this disorder?
POLG (nuclear DNA);
no (highly variable)
Name five syndromes associated with mutations in mtDNA.
(the first two are acronyms)
(the last one (LS) is usually associated with mutations in nuclear DNA)
MELAS;
MERRF;
Kearne-Sayre syndrome;
Pearson syndrome;
Leigh syndrome (although most cases come from mutations in nuclear DNA)
MELAS is a disorder that results from mutations in mtDNA.
What does MELAS stand for?
Is it hetero- or homoplasmic?
(What are some of its signs and symptoms?)
Myopathy, mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like episodes’
heteroplasmic;
(weakness, ataxia, headache, seizures, dementia, vomiting, transient hemiparesis, lactic acidosis, deafness)
MERFF is a disorder that results from mutations in mtDNA.
What does MERFF stand for?
Is it hetero- or homoplasmic?
(What are some of its signs and symptoms?)
Myoclonic Epilepsy with Ragged Red Fiber;
heteroplasmic;
(Myopathy, ataxia, sensorineural deafness, dementia)
Kearne-Sayre syndrome is a disorder that results from mutations in mtDNA.
Is it caused by genetic duplications, deletions, or point mutations?
What are some of its signs and symptoms (don’t memorize, just be aware of some of these options)?
Deletion;
progressive external ophthalmoplegia of early onset, cardiomyopathy, heart block, ptosis, retinal pigmentation, ataxia, diabetes
Pearson’s syndrome is caused by what kind of genetic mutations?
What are two of its signs or symptoms?
Deletions in mtDNA;
sideroblactic anemia, exocrine pancreatic dysfunction
Leigh’s syndrome is caused by what kind of genetic mutations?
What are two of its signs or symptoms?
Deletions in mtDNA;
lesions in the basal ganglia and midline brain stem
Kearne-Sayre syndrome, Pearson’s syndrome, and Leigh’s syndrome are all caused by what sort of genetic mutation?
Deletions in mtDNA
How do lysosomal enzymes act upon macromolecules within the lysosome?
In a step-wise fashion
(one enzyme after the other in a specific order)
True/False.
If a single step in the normal lysosomal enzyme step-wise pathway is missing, all subsequent steps will likey not occur.
True.
Most lysosomal storage diseases follow what inheritance program?
Autosomal recessive
(mutations in single enzymes)
True/False.
Lysosomal storage diseases typically present in newborns.
False;
these are chronically progressive diseases that present with normal newborns
What are the five main categories of lysosomal storage disorder?
(note: they are categories named for the lysosomal substrate involved)
Mucopolysacharridoses;
sphingolipidoses;
glycogen storage;
glycoproteinoses;
mucolipidoses
(also membrane transporter defects)
How do amino acids, carbohydrate monomers, and small lipids leave lysosomes after being digested out of larger structures?
There are many specific transporters in the lysosomal membrane for these building block units
What disease is an example of a disorder resulting from lack of a certain amino acid transporter in the lysosomal membrane?
What amino acid is it that cannot leave and crystalizes / builds up in the cell?
What is the result?
Cystinosis;
cystine;
cell death
Lamellar, circular lysosomes are associated with what type of disorder?
Sphingolipidoses
What are some of the major first signs (in general) of a lysosomal storage disease in terms of noticing something in a child that has met their cognitive and motor milestones?
School failure, regression, seizures
Mucopolysaccharidoses involve a failed lysosomal digestion of:
Glycosaminoglycans (GAGs)
Name a few major sphingolipidoses.
Tay-Sachs disease;
Gaucher’s disease;
Fabry’s disease;
Niemann-Pick disease
What is the most common form of lysosomal storage disease?
What substance is building up in the lysosome in this condition?
Gaucher disease;
glucocerebroside
What organs are most affected in Gaucher disease?
What enzyme is deficient?
Bone, liver, spleen;
glucocerebrosidase
The following are all examples of what form of lysosomal storage disease?
Tay-Sachs disease
Gaucher disease
Fabry’s disease
Niemann-Pick disease
Sphingolipidoses
What are Gaucher cells?
What organs are typically swollen in Gaucher disease?
Lipid-laden macrophages;
liver and spleen
A mother brings in her child with hepatosplenomegaly.
Through lab work, lipid-laden macrophages are identified.
What is the likely diagnosis?
Gaucher disease
How can lysosomal storage diseases be treated?
IV infusions of the missing enzyme every 1-2 weeks
What type of disorder results due to a failure of attachment of mannose-6-phosphate to certain enzymes?
To where would M-6-P target these enzymes?
I-cell disease;
lysosomes
How does I-cell disease work?
A lack of mannose-6-phosphate means lysosomal enzymes are exocytosed
What organ is typically hit first in Fabry disease?
What type of lysosomal storage disease is Fabry disease?
What are the initial symptoms?
The glomeruli;
a sphingolipidosis;
severe rash, proteinuria, corneal whorl clouding
What are the general signs seen in mucopolysaccharidoses (a type of lysosomal storage disorder)?
Bone/liver/spleen issues;
coarsening of the face;
corneal clouding
What are two examples of a mucopolysaccharidosis with somatic and neurologic effects?
These are a type of:
Hurler’s (type I), Hunter’s (type II);
lysosomal storage disorder
What is an example of a mucopolysaccharidosis with only somatic effects?
What is an example of a mucopolysaccharidosis with only neurologic effects?
These are a type of:
Morquio syndrome (type IV);
Sanfilipo syndrome (type III);
lsosomal storage disease
What are a few examples of glycogen storage diseases (a type of lysosomal storage disease)?
von Gierke’s disease;
Pompe’s disease;
Cori’s disease;
McArdle’s disease
Ataxia telangiectasia is associated with mutations on what gene?
Ataxia telangiectasia is associated with what type of tumor?
Ataxia telangiectasia is associated with a deficiency of what antibody?
XTNR, ATM;
ocular angiomas;
IgA
What are the common signs / symptoms of Sturge-Weber syndrome?
(facial, ocular, CNS (x3))
Facial port-wine stains,
glaucoma,
seizures,
intellectual disability,
ipsilateral leptomeningeal angioma
Name two lysosomal storage diseases that are X-linked recessive.
Fabry’s;
Hunter’s
What are the major signs of Menke’s disease?
Short stature;
white-gray, kinked hair
- One of the commonest LSDs which is inherited in an X-linked fashion is Fabry Disease, which causes proteinuria and renal disease. An unusual presenting feature of Fabry Disease is:
A. mental retardation
B. hepatomegaly
C. pathologic fractures
D. corneal whorl opacification
E. congenital malformations
D. corneal whorl opacification
- A mucopolysaccharidosis condition which manifests with neurologic regression and skeletal signs of storage and hepatosplenomegaly (neurologic + somatic features) in early childhood is _____.
A. MPS I (Hurler)
B. MPS III (Sanfilipo)
C. MPS IV (Morquio)
D. Tay-Sachs
E. cystinosis
A. MPS I (Hurler)
- A child is born with stenosis of pharyngeal nostrils (choanal atresia), a coloboma of the iris and and malformations of the ear. The chromosomal evaluation is normal and there are no obvious prenatal or family history features which explain this set of abnormalities. Which of the following is the likely diagnosis?
A. VATER association
B. CHARGE association
C. Maternal PKU effects
D. Fetal alcohol syndrome
E. Oligohydramnios sequence
B. CHARGE association
What neurological sequelae are seen in Fabry disease?
None
- In children with inborn errors of metabolism such as isovaleric academia, a metabolic exacerbation leading to acidosis and illness can be caused by which of the following?
A. change in weather
B. intercurrent illness (colds) causing catabolism
C. puberty
D. emotional stressors
B. intercurrent illness (colds) causing catabolism
- Signs of a neurocutaneous disease such as NF1 and tuberous sclerosis include numerous clinical signs. In addition to clinical signs, which of the following is helpful in making a diagnosis of either condition?
A. chromosomal analysis
B. positive family history in a parent
C. microcephaly
D. limb reduction abnormality
B. positive family history in a parent
- Metabolic inherited diseases are often ascertained by newborn screening. The abnormality detected in NBS testing in a newborn with PKU is which of the following?
A. elevated phenylalanine in dried blood spots
B. chromosomal abnormality
C. urine phenylketones
D. elevated fatty acids in dried blood spot
A. elevated phenylalanine in dried blood spots
- In diagnosing a child with disproportional short stature, which of the following tests is most useful?
A. chromosomal analysis
B. urine organic acid analysis
C. complete skeletal series
D. bone marrow biopsy
C. complete skeletal series
- Mitochondrial disorders often do not appear to follow obvious Mendelian inheritance patterns. In conditions such as MELAS, which is the typical inheritance pattern?
A. multifactorial inheritance
B. autosomal dominant with decreased penetrance
C. sporadic inheritance
D. maternal inheritance
D. maternal inheritance
- Diagnosis of neurofibromatosis 1 (NF1) is based on the presence of two or more specific criteria. Which one of the following criteria is NOT included in the NIH Guidelines for clinical diagnosis of NF1?
A. Reduced levels of RAS expression
B. Freckling in the armpit or groin area
C. Two or more neurofibromas
D. Optic glioma
E. Family history of NF1
A. Reduced levels of RAS expression
- 7 year old child is seen by the neurologist because of the combination of multiple café au lait macules, Lisch nodules, and freckling in her armpits and groin. She is MOST at risk for developing:
A. aortic dilatation
B. neurofibromas
C. polycystic kidneys
D. cardiomyopathy
E. neurologic regression
B. neurofibromas
- Which one of the following statements about genetic defects in oxidative phosphorylation is true?
A. All genetic defects of oxidative phosphorylation follow an autosomal recessive pattern of inheritance.
B. All genetic defects of oxidative phosphorylation follow a maternal pattern of inheritance.
C. Genetic defects of oxidative phosphorylation generally affect tissues and organs that rely on glycolysis as the sole energy source (e.g. red blood cells). D. Genetic defects of oxidative phosphorylation are due to mutations in the mitochondrial genome in majority of cases.
E. Some genetic defects of oxidative phosphorylation follow an autosomal recessive pattern of inheritance and some follow a maternal pattern of inheritance.
E. Some genetic defects of oxidative phosphorylation follow an autosomal recessive pattern of inheritance and some follow a maternal pattern of inheritance.
A deficiency of the enzyme α-galactosidase A leads to what disease?
Note: copied from Brainscape Step 1 flashcards.
Fabry’s disease
A deficiency of what enzyme leads to what Fabry’s disease?
Note: copied from Brainscape Step 1 flashcards.
α-galactosidase A
What type of inheritance does Fabry’s disease demonstrate?
Note: copied from Brainscape Step 1 flashcards.
X-linked recessive
A patient has peripheral neuropathy of the hands and feet, angiokeratomas of the skin, and both cardiovascular and renal disease.
These characteristics are typical of which lysosomal storage disease?
Note: copied from Brainscape Step 1 flashcards.
Fabry’s disease
What are some of the main organ systems affected by Fabry’s disease?
PNS (peripheral neuropathy of the hands and feet),
skin (angiokeratomas),
heart,
kidneys
What substrate accumulates in a patient with a deficiency of α-glucocerebrosidase?
Where?
Note: copied from Brainscape Step 1 flashcards.
Glucocerebroside;
the liver, spleen, and bone (Gaucher’s disease)
A child has progressive neurodegeneration, developmental delay, cherry-red spots on the macula, and lysosomes with onion skinning; he does not have hepatomegaly. These characteristics are typical of which lysosomal storage disease?
Note: copied from Brainscape Step 1 flashcards.
Tay-Sachs disease
(hepatomegaly here would indicate Niemann-Pick)
What sign is useful in distinguishing between Tay-Sachs and Niemann Pick?
Hepatomegaly (present in N-P)
What enzyme deficiency leads to Gaucher’s disease?
α-Glucocerebrosidase
What is the most common lysosomal storage disorder?
Gaucher’s disease
A patient has developmental delay, gargoylism, airway narrowing, and corneal clouding. These characteristics are typical of which lysosomal storage disease?
What is the inheritance pattern?
Hurler’s syndrome;
autosomal recessive
For which three lysosomal storage disorders are Ashkenazi Jews at an especially increased risk?
Tay-Sachs, Niemann-Pick, and some forms of Gaucher’s disease
Gaucher’s disease inheritance?
Autosomal recessive
What are Gaucher cells?
Macrophages that look like crumpled tissue paper on microscopy (they are full of glucocerebroside)
Niemann-Pick disease is a deficiency of:
What inheritance pattern does it follow?
Sphingomyelinase;
autosomal recessive
A child has progressive neurodegeneration, hepatosplenomegaly, and a cherry-red spot on his macula. These characteristics are typical of which lysosomal storage disease?
Note: copied from Brainscape Step 1 flashcards.
Niemann-Pick disease
Cherry-red spot on macula + hepatosplenomegaly = probably _________________
Cherry-red spot on macula w/o hepatomegaly = probably _________________
Niemann-Pick’s disease;
Tay-Sachs disease
What type of inheritance does Niemann-Pick disease demonstrate?
Autosomal recessive
What enzyme deficiency leads to Tay-Sachs disease?
Hexosaminidase A
A deficiency of hexosaminidase A leads to:
Inheritance pattern:
Tay-Sachs disease;
autosomal recessive
What enzyme is deficient in Tay-Sachs disease?
What substance accumulates?
Hexosaminidase A;
GM2 ganglioside
What two lysosomal storage diseases may present with a cherry-red spot on the macula?
Niemann-Pick disease;
Tay-Sachs disease
What substrates accumulates in Hurler’s syndrome?
What pattern of inheritance is shown?
Heparan sulfate and dermatan sulfate;
autosomal recessive
What substrates accumulates in Hunter’s syndrome?
What pattern of inheritance is shown?
Heparan sulfate and dermatan sulfate;
X-linked recessive
A patient has mild developmental delay, gargoylism, and airway obstruction but no corneal clouding. These characteristics are typical of which lysosomal storage disease?
Note: copied from Brainscape Step 1 flashcards.
Hunter’s syndrome
What the two lysosomal storage diseases lead to an accumulation of heparan sulfate and dermatan sulfate?
Hunter’s and Hurler’s
What two lysosomal storage disorders shown an X-linked recessive pattern of inheritance?
Fabry’s disease;
Hunter’s syndrome
What enzyme is deficient in Krabbe’s disease?
What substrate accumulates?
What is the inheritance mode?
Galactocerebrosidase;
galactocerebroside;
autosomal recessive
Which type of mucopolysaccharidosis shows only somatic symptoms?
Which type of mucopolysaccharidosis shows only neurologic symptoms?
Which types of mucopolysaccharidosis show both neurologic and somatic symptoms?
Morquio’s;
Sanfilipo’s;
Hunter’s and Hurler’s
Which type of mucopolysaccharidosis shows only somatic symptoms?
(no neurologic)
Marquio’s
Which types of mucopolysaccharidosis show both neurologic and somatic symptoms?
Hunter’s and Hurler’s
Which type of mucopolysaccharidosis shows only neurologic symptoms?
(no somatic)
Sanfilipo’s
What are the ‘vital signs’ of dysmorphology?
Height, weight, and head circumference
Of newborns with structural defects, about 1/4 have multiple anomalies.
What is the singular leading cause of multiple anomalies in a newborn?
(note: only 50% of cases with multiple anomalies even have a known etiology)
Chromosomal abnormalities
(76% of known etiologies)
Do singular anomalies in newborns typically have an identifiable etiology?
No;
they are typically multi-factorial
Define ‘sequence’ in terms of dysmorphology and congenital defects.
One anomaly leads to other developmental anomalies
(e.g. Potter’s syndrome)
What is the acronym for the signs of Potter’s syndrome?
POTTER
Pulmonary hypoplasia
Oligohydramnios
Twisted face
Twisted skin
Extremity abnormalities
Renal agenesis
Define ‘dysplasia’ in terms of dysmorphology and congenital defects.
Intrinsic developmental abnormality due to defective cellular organization
(e.g. skeletal dysplasia;
effects of fetal alcohol syndrome, thalidomide)
Define ‘disruption’ in terms of dysmorphology and congenital defects.
Interruption of normal morphological effects due to an extrinsic force
(e.g. amniotic band syndrome)
Define ‘deformation’ in terms of dysmorphology and congenital defects.
A change of normal morphology caused by extrinsic forces
(e.g. clubfoot)
Define an ‘association’ disorder in terms of dysmorphology and congenital defects.
An exclusion diagnosis of a nonrandom pattern of effects
(e.g. VACTERL, or CHARGE)
Name each type of dysmorphology illustrated by the separate examples below:
Amniotic band syndrome
Potter’s syndrome
Clubfoot
Amniotic band syndrome - disruption
Potter’s syndrome - sequence
Clubfoot - deformation
Name each type of dysmorphology illustrated by the separate examples below:
Fetal alcohol syndrome
Skeletal dysplastic syndromes
VACTERL syndrome
Fetal alcohol syndrome - dysplasia
Skeletal dysplastic syndromes - dysplasia
VACTERL syndrome - association
True/False.
Facial dysmorphological changes can often predict location changes and problems in the brain.
True
(“the face predicts the brain)
(e.g. a child with a midline cleft lip / palate might also have holoprosencephaly)
What are the two steps of clinical research that happen before human test subjects are involved?
Basis - lab work to establish potential therapy basis
Pre-clinical - synthesis, toxicology, formulation, regulatory
Phase I studies use a small group of healthy individuals to do what?
Phase II studies do what?
Establish safe dosage and schedule for new therapy;
establish biological effect, establish efficacy
Phase III studies do what?
Phase IV studies do what?
Compare new therapy to old therapy;
figure out where the new therapy can fit into the current market
In phase I trials, we say: “is it _________?”
In phase II trials, we say: “is it _________?”
In phase III trials, we say: “is it _________?”
In phase IV trials, we say: “is it _________?”
Safe
Efficacious
better than the existing therapy
market-ready
What four disorders comprise almost 70% of all skeletal dysplasias?
Achondroplasia,
thanatophoric dysplasia,
osteogenesis imperfecta,
achondrogenesis
What is the very severe skeletal dysplasia in which cartilage is not produced?
This condition is often lethal due to a narrow thorax and pulmonary hypoplasia.
Achondrogenesis
What disorder is characterized by a ‘clover-leaf’ skull, frontal bossing, midface hypoplasia, micromelia, macrocephaly, brachydactyly, and CNS abnormalities?
What is the inheritance pattern and associated gene?

Thanatophoric dysplasia
Autosomal dominant - FGFR3 gene
What is the most common cause of disproportionate short stature?
Achondroplasia
In what fairly common skeletal dysplasia is there sometimes craniocervical junction compression, increasing the risk of death in infancy?
Achondroplasia
In addition to achondroplasia, what other two disorders are linked to mutations in the FGFR3 gene?
Thanatophoric dysplasia,
hypochondroplasia
Name some of the signs associated with thanatophoric dysplasia.
‘Clover-leaf’ skull, frontal bossing, midface hypoplasia, micromelia, macrocephaly, brachydactyly, and CNS abnormalities
Which tends to be more severe in its skeletal features, achondroplasia or hypochondroplasia?
Which tends to show intellectual disability and/or epilepsy more often?
Achondroplasia;
hypochondroplasia
What type of osteogenesis imperfecta is characterized by blue sclerae, fragile bones, triangular face, and (sometimes) deformed teeth?

Type I
(Note: multiple fractures in various stages of healing)

What type of osteogenesis imperfecta is a ‘progressively deforming’ type?
Type III
What type of osteogenesis imperfecta is similar to type I but is not characterized by blue sclerae?
Type IV
What type of osteogenesis imperfecta is often lethal at or near birth?
Type II
The ratio of upper to lower body segment is a way of confirming proper limb length.
What ratio is normal at birth?
What ratio is normal ≤ 10 years of age?
What ratio is normal after > 10 years of age?
1.7
1
< 1
(a higher number may indicate short extremities)
What should arm span typically be approximately equal to?
Height
Why is achondrogenesis a lethal disorder?
Failure of cartilage growth = no endochondral ossification = no long bones

What are some types of skeletal dysplasia that are often lethal in the fetal or perinatal stage?
Achondrogenesis
Thanatophoric dysplasia
Asphyxiating thoracic dysplasia
Osteogenesis imperfecta type II
What shape is the skull in thanotophoric dysplasia?
What shape is the thorax?
Clover-leaf shaped;
bell-shaped
The achondroplastic mutation in FGFR3 is a _____________ mutation.
gain-of-function
(FGF receptor 3 supresses bone growth)
FGF receptor 3 has what effect on bone growth?
How does this play into achondroplasia?
It suppresses bone growth;
the FGFR3 mutation seen in achondroplasia is a gain-of-function mutation
What disorder presents very similarly to achondroplasia but is a result of mutation in the cartilage oligometric protein gene (COMP)?
Pseudoachondroplasia
The clinical heterogeneity of osteogenesis imperfecta is explained by which, allelic or locus heterogeneity?
Both
In type I osteogenesis imperfecta, is the problem of type I collagen quality or quantity?
Quantity
What are the general requirements for sensitivity and specificity of a screening test?
Sensitivity as high as possible (~0 false negatives);
acceptably high specificty
Phenylketones are produced in high number when what substance is found in high concentrations in the body?
What enzyme is likely absent?
Phenylalanine;
phenylalanine hydroxylase
Phenylalanine hydroxylase turns phenylalanine into:
Tyrosine
How is PKU screening done today?
Mass spectrometry
(extremely accurate)

Individuals with phenylketonuria have excessively high levels of _________ and low levels of _________.
Phenylalanine;
tyrosine
What are the three components of PKU treatment?
Phenylalanine restriction (but not complete removal!);
tyrosine supplementation;
tetrahydrobiopterin (BH4) supplementation
Why do individuals with PKU often have a ‘musty’ smell?
Why do individuals with PKU often have lightly colored skin and hair?
Phenylketone abundance;
tyrosine deficiency (a precursor to melanin)
Why is it important that metabolic disorders be screened for in newborns?
Proper nutrition + medication of these infants can often save their lives / neurological capacity
What cofactor is necessary for proper phenylalanine hydroxylase function?
Tetrahydrobiopterin
(BH4)
What is a confounding factor that makes the diagnosis of acute metabolic diseases of infancy difficult to diagnose?
They are clinically indistinguishable from sepsis
What are the signs and symptoms associated with the VACTERL diagnosis?
Vertebral anomalies;
anal atresia;
cardiac anomalies;
tracheo-esophageal fistulas;
renal anomalies;
limb deformities
What are the signs and symptoms associated with the CHARGE diagnosis?
Coloboma,
heart anomalies,
atresia choanae,
growth retardation, genital abnormalities,
ear abnormalities.
In diagnosing a child with disproportional short stature, which of the following
tests is most useful?
A. chromosomal analysis
B. urine organic acid analysis
C. complete skeletal series
D. bone marrow biopsy
C. complete skeletal series
A newborn baby is very ill and has a distinctive ‘sweaty socks’ smell.
Assuming this comes from an inborn error of metabolism, what disorder is responsible for the smell and illness?
Isovaleric acidemia
