Cancerogenes Flashcards
Varför har cancerfall per år ökat från 70-talet och framåt?
Hur många få cancer under sin livstid?
- Delvis beroende på ökad medellivslängd, dock inte hela förklaringen
- 1/3 får cancer under livet
Varför finns det kulturella skillnader gällande cancertyper ho befolkningen?
- Beror mkt på kulturella skillnader, ex betelnötter i Asien (cancerframkallande i esofagus)
- Spegling av riskfaktorer i dessa områden

Hur kan vi se att magsäckscancern i Japan främst beror på miljlön och hereditet (nutrition, hårt grillad fisk ex skapar cancerframkallande ämnen vilket tas upp i lever)
Migrationsstudier
- Hos japaner som flyttat till USA går frekvens av lever/magsäcks-cancer ner
- Nästa generation fortsatte tendensen

- Lungcancerökning hos manliga engelska läkare, går direkt relatera till ökad rökning
- Hos kvinnliga läkare ses en långsammare ökning av rökning varpå det ses långsammare ökning av lungcancer
- Varför kan vi kalkylera att kvinnors utveckling av bröstcancer i lungor kommer öka?
Latenstid från exponering till drabbning

Vad kan vi dra för slutsats av att 85-90 % av all cancer har ursprung i de livsfaktorer som vi exponeras för (kemikalier, strålning, virus, bakterier osv), det vill säga, vad innehåller de?
Att cancerceller innehåller somatiska mutationer och/eller epigenetiska (förändring i hur gener uttrycks) t ex
Hur kan ett främmande exogent ämne framkalla cancer?
- Oftast fettlösliga, behöver därför inga transportproteiner
- Diffunderar in i membranet enkelt
- Kemisk struktur känns igen av proteiner/enzymer vilket i bensens fall gör att det kommer metaboliseras till en molekyl med OH-grupp och sedan kan det ske vidare konjugeringar och den blir mer vattenlöslig och kan skickas ur cellen på nytt
- Men sker ofta reaktiva mellansteg där de kan reagera med proteiner/DNA vilket kan bilda kemiska föreningar (kovalent bundna metaboliter i DNA) vilket i nästa cell kan ge mutation

Varför borde du sluta äta jordnötter?
- Jordnötter som lagras fel i varma länder t ex bildar aflatoxin som
- Metaboliseras och bildar epoxid som binder in till DNA-molekyl och bildar mutationer
- Krävs bara små mängder för att utveckla cancer
På vilket sätt gör miljöfaktorer och genetisk konstitution att vi utvecklar cancer?
Samspel

Vilka är de två viktigaste Hallmarks of cancer och hur förhåller sig deras genes gällande dominant/recessiv?
-
Oncogener (gas) – Kan aktiveras t ex om mutation eller förändrad genexpression (ex en epigenetisk förändring, förändring av genuttrycket) – ger gain of function
- Dominant (räcker på ena genen (mors eller fars))
-
Suppresorgener (broms) – ger negativa signaler och minskar hastighet av proliferation
- Recessiv
Hur fungerar care takers och vad händer när de muteras?
Den tredje Hallmarken
- DNA-reparationsgener
- Håller genomet stabilt, undviker mutationsgener
- Mutationer på dessa gör att de reparerar sämre varför andra mutationer ökar
Nämn minst tre generella sätt som en ONCO-gen kan ge GAIN OF FUNCTION?
- Kan ske genom translokation av genen (överkorsning av kromosomerna), kan därför hamna på ny kromosomal plats, erhåller då t ex ny promotor (den nya promotorn stimulerar kanske i högre utsträckning vilket ger mkt mer av proteinet), gendosökning liksom (detta är ingen mutation)
- Vid celldelning kan amplifiering ske (flera kopior av genen) vilket gör att det produceras mer protein
- Även förändringar som ligger utanför genen, t ex promotorn vilket ökar transkriptionen av genen (mutation av promotorn), vilket ökar mängden protein
- mutation i proteinet (ex en nukleotidförändring) vilket gör att koden förändras och vi får ett annat protein

Vilka generella procancer-egenskaper kan ett somatiskt muterat protein få?
- Bryts inte ner lika lätt
- Mer effektiv (hyperaktivt protein) som kan stimulera ex tillväxt (inte mer av proteinet men snabbare liksom)
Vad är det mest sannolika resultatet av ett slumpvis somatiskt muterat protein?
Ett sämre protein vilket inaktiverar (likviderar) den muterade genen

Translokation av ONCO-genen c-myc är vanligt förekommande vid cancer, vad händer när detta sker?

- Igh-promotorn kontrollerar då uttrycket av c-myc som ökar
När MYC ökar i koncentration tränger den bort fler av MAX-proteinerna vilket ger en MYC-MAX heteromer som binder till DNA och stimulerar/aktiverar transkription av gener som är intressanta för cellcykeln och cellproliferation/tillväxt

Philadelphiakromosomen bildas efter en translokering där det börjar kodas för ett protein som ses vid kronisk myoelid leukemi.
Vilken Hallmarkegenskap har detta protein och vad är det som sker?
ONCO-gen
- Genen ABL (från kromosom 9) translokeras till kromosom 22
- ABL-genens axon hamnar tillsammans med BCR-gen vilket ger ett fusionsprotein, en del BCR och en del ABL
- som är ett ett kinas som stimulerar fosforylering och signalvägar i cellens blodceller (vita blodkroppar) och ger proliferation och bildning av yolid arm
- Sker i blodets stamceller i benmärg
- För att bota måste stamceller i benmärgen strålas så att dessa celler dödas av och sedan individ transplanteras med vita blodkroppsproducerande celler

RAS är en viktig ONCO-gen och sitter intracellulärt, precis innanför membran, när tillväxtfaktor binder leder det till olika fosforyleringar som i slutändan stimulerar transkriptionsfaktorer. Hur kan den muteras och vad blir dess resultat?
- Är en ONCO-gen, kan muteras i tre olika positioner som håller GTP
- Vanlig mutation på aminosyra 12 vid cancer där glycin byts till valin vilket gör att GAP inte kan inaktivera RAS vilket gör signalvägen ständigt aktiv (fastnar i ON-state)
Missense-mutation i antingen kodon 12, 13 eller 61 aktiverar RAS proteiner
(RTK = receptor tyrosinkinas

Vad är fosfatidylinositol-3-kinas (PIK3CA) vanliga funktion?
- Tillväxtfaktorer binder till receptor tyrosinkinas vilket ger autofosforylering av proteiner och PIK3CA (P110 och p85)
- Som också överför fosfotgrupper (men inte till protein) till lipiden PIP3 (tre fosfatgrupper vid aktivering)
- Denna kan användas av andra kinaser för att aktivera AKT som kan aktivera en mängd vägar; reparation, cellcykelstopp, apoptos mm.

Vilken Hallmark-typ är PIK3CA?
ONCO-gen
Hur muterar PIK3CA som cancergen och vilken effekt får den?
- Specifika positioner på PIK3kinaset kan muteras och ge mer fosforylering av PIP3 vilket stimulerar fosforylering av AKT som aktiverar en mängd vägar

Vad gör PTEN?
- PTEN (fosfatas), tar bort fosfatgrupper från PIP3 och minskar aktiveringen PIK3CA– Tumörsuppresor

Tumörsuppressorgener
- Hämmar eller motverkar cellproliferation
- Funktion som försvinner (därför svårare att identifiera)
- Recessiv
- Familjära fall (dvs att risken att utveckla cancer i retina fanns i familjen men också sporadiska fall utan denna härledning)
- I de familjära fallen fanns mutationen i en av allelerna av föräldrarna som ärvdes av barnet, en muterad och en frisk allel som kan upprätthålla funktion, men om retina var känslig för ytterliggare mutatin som inaktiverar andra allel så försvinner bromsande funktionen av proliferationen
- I de sporadiska fallen fick pat. först en mutation i en cell och senare en andra mutation för att inaktivera bägge celler vilket är sällsynt
- Situationen ovan är vanligare
Vad kallas denna teori för?
Kallas för second hit då första hiten givits av föräldrar

Tumörsuppressorgenen RB är den första upptäckta tumörsuppressorn och inaktiveras helt (oftast) enligt second hit-teorin.
Hur verkar den i vanliga fall?
- Aktiv vid restriktionspunkten i cellcykeln
- När cellen får stimulering (signaler utifrån) om celldelning, associerar cyklin D och cyklin D beroende kinas och fosforylerar retinoblastomprotein (RB) innan S-fas
- I vanliga fall binder RB upp transkriptionsfaktorn E2F och fosforyleringen leder till att RB släpper iväg E2F för ”lagom stimulering” och cellcykeln kan gå in i S-fas

Tumörsuppressorgenen RB är den första upptäckta tumörsuppressorn och inaktiveras helt (oftast) enligt second hit-teorin.
- Genen är i vanliga fall aktiv vid restriktionspunkten i cellcykeln
- När cellen får stimulering (signaler utifrån) om celldelning, associerar cyklin D och cyklin D beroende kinas och fosforylerar retinoblastomproteinprotein (RB) i G1
- RB binder upp transkriptionsfaktorn E2F och fosforyleringen leder till att RB släpper iväg E2F för ”lagom stimulering” och vidare push till S-fas
Hur verkar den när den är muterad?
- När RB (tumörsupressor) är muterat, binder den inte E2F, de förblir separerade vilket lämnar E2F fritt att stimulera t ex replikation
- Nedärvd mutation gör att cellproliferation stimuleras hela tiden, framför allt då om vildtyp är förlorad ”second hit” - retinoblastom

p53 agerar tumörsuppressor, hur agerar den i normalfallet vid stress (DNA-skada, hypoxi etc)?
- Då aktiveras protein ATM (kinas) som fosforylerar p53 som släpper från sitt hämmande protein MDM2 och kan binda DNA för att stimulera transkription som p21 som hämmar cyklin D så det inte kan fosforylera RB och därmed inte släppa E2F vilket ger ett cellcykelstopp innan S-fas
- Då får cellen tid för DNA-reparation/eliminera olika stressfaktorer som hydroxiradikaler osv, om cellen inte lyckas kan p53 transkribera apoptosgener – bättre att cellen dör än att dra på sig mutationer
- MDM2 binder i vanliga fall upp p53 och stimulerar till ubiquitinering och degradering av proteasom
- p53 reglerar också MDM2 (negativ feedback) så cellens nivåer av P53 hålls i normalfallet låga
- p53 agerar också i G2 innan M-fas

Vad kan sker vid en procancer-mutation av p53?
Vid mutation av p53 påverkar det ofta kontaktytorna med DNA-molekylen så att p53 inte kan utöva verkan (reparation/apoptos/produktion av MDM2 osv) och cellen passerar G1 och prolifererar mer
Vad visar bilden?

- Bilden visar att homozygota med en viss mutation på p53 drabbas hårt men att även de heterozygota är predisponerade att utveckla cancer vid ”rätt” miljöbetingelser eftersom p53 normalt har funktion att avstanna cellcykeln, stimulera DNA reparation och inducera apoptos vid cellulär stress och DNA-skador
På vilket sätt är det vanligast att p53 förlorar sin verkan?
Vanligast är att den normala kopian förloras via endogena mekanismer (s.k. ”loss of heterozygosity”), vid mitosen blir det fel när kromosomerna skall fördelas på dottercellerna, ena dottercellen får tre kopior, den andra får endast en kopia. Om den cellen då förlorat sin normala kopia, finns bara den muterade kopian kvar och genen har i den cellen förlorat sin funktion, dvs. båda allelerna har inaktiverats. Cellen blir “homozygot” med avseende på den muterade genen (Knudson´s ”two-hit hypothesis”)
- Sannolikheten att förvärva en ”andra hit” är så stor att den troligen inträffar under en livstid och man utvecklar en cancer, dvs. nedärvningsmönstret av fenotypen cancer ser dominant ut.
- Vid cancer utan ärftlig betingelse är mutationer i detta protein vanligt (50 %)
Vilket protein pratar vi om?
p53
Vad gör care takers och vad händer när de blir muterade och tappar funktion?
Ge ett exempel
- DNA-reparationsgener, korrigerar ex mutationer/skador
- Mutation här gör att andra mutationer inte repareras (ONCO, tumörsupressorer)
- Ett exempel är UV-strålning som ger mutationer i DNA, förändrar strukturen på dubbelspiralen, DNA-reperationssystem avlägsnar förändring (t ex tymin-tymindimer), en DNA-sträng utgör mall och korrekta baser kan fyllas i, finns inte detta system får personer som ärvt detta genetiskt cancer
Vissa gener är kritiska för cancerutveckling (RAS, p53, PIK3CA etc), vad kallas de för?
Drivers
Hallmarks of cancer
ONCOgener står för gasen och supressorn för bromsen, oftast räcker inte dessa för att omvandla normal cell till tumörcell utan kräver också
- Okänslighet för apoptosignaler
- Angiogenes
- Obegränsat antal replikeringar
- Förmåga att invadera/metastasera normal och annan vävnad (det som dödar)

- Mutationer kan också uppstå under den högproliferativa processen kan bidra till tumörprogressionen men behöver inte göra det
Vad kallas de för?
Passangers
Vilka två teorier om klonal utveckling finns?
-
Klonal evolutionsmodel
- Initial mutation, tillväxt av cellen och dess dotterceller med mutationen, byggs på med fler mutationer och ytterligare skjuts av proliferation och fler mutationer vilket ger heterogenitet i sluttumören
-
Cancerstamceller
- Stamceller som muterar och ger upphov till tumörceller som växer snabbare än normala celler, detta ger också normalceller och cancerceller
- Dessa ger en heterogenitiet av olika grad och modellerna kompletterar eventuellt varandra (evolution och cancerstamceller)

Vad visar bilden?

- Första driver-mutation tidigt, olika kloner med tumörceller där den ursprungliga drivern finns kvar men också kloner av tumörceller där mutationen inte finns kvar och andra typer av mutationer (passengers)
Coloncancer finns i en familjär form som heter FAP
- APC (tumörsupressor) upprätthålls om inte bägge alleler är påverkade
- Vid mutation av APC växer det snabbare (bildar tusentals polyper/adenom), det bildas fler mutationer med snabbare växt och fler mutationer… carcinom bildas till slut
Vilken funktion har APC vid normaltillstånd och vad händer när det är muterat?
- Normal funktion för APC-genen är att ingå i ett proteinkomplex (hjälper till att hålla ihop komplexet), kinas i komplexet fosforylerar betakatenin, som då ubiqutineras och degraderas i proteasom
- Vid inbindning av tillväxtväxtfaktor inaktiveras kinas vilket gör att betakatenin kan stimulera transkription av tillväxt osv
- När APC är muterat håller inte APC inte ihop komplexet och betakatenin fosforyleras inte, därför stimulerar betakatenin proliferation även utan initial tillväxtfaktor

Telomeras är ett protein som har förmågan att förlänga telomerfunktionen, cellen kan leva fler cellgenerationer
- Ej funnit mutationer i telomerasgenen
- Fungerar som ONCO-gen då det uppreglerar
Så vad har man funnit?
Mutationer i promotorn till telomerasgenen som gör att transkriptionsfaktorer kan stimulera till mer telomerasmolekyler som då bygger upp telomererna vilket förklarar dess ökning i t ex hjärntumörer
Angiogenes är viktigt för att transportera in syre och näring till tumören, skulle begränsa tumörens storlek avsevärt utan angiogenes.
Vad sker i normalfallet vid god tillgång på O2 eller sämre tillgång på O2 med HIF-1-alfa?
- Proliner på HIF-1-alfa kommer att hydroxileras av prolylhydroxilaser (som kräver alfa-ketoglutarat), dessa hydroxylgrupper är bindningsplatser för ubiqitinering som sätts på och HIF-1-alfa degraderas i proteasom
- Vid för lite O2 sker inte hydroxylering varpå HIF-1-alfa binder till HIF-1-beta i cellkärnan, fungerar då som transkriptionsfaktorer för att korrigera syrebristen, ex för kärlbildning, cytoskelettstruktur, cellproliferation, cellöverlevnad, glukosmetabolism

Vad sker vid mutering av HIF-gener (minst 3 finns som är cancerframkallande)?
- Den kommer fortsätta stimulera då det inte degraderas
- HIF-1-alfa binder till HIF-1-beta i cellkärnan, fungerar då som transkriptionsfaktorer
- ex för kärlbildning, cytoskelettstruktur, cellproliferation, cellöverlevnad, glukosmetabolism

- Tumören ska växa igenom membran (svårt, men kan ske genom uppreglering av proteaser, kollagenaser) och sedan ta sig in i blodkärl (egentligen ganska oxidativ miljö och svårare att överleva) och lymfkärl (mer aktiva här) var de släpper från modertumör, kan fastna i finare kapillärnät/lymfa,
- Krävs vissa egenskaper hos vävnaden där den fastnar, måste få de näringsämnen och syretillförsel den kräver (svårt)
Vad kallas denna teori för?
Seed and Soil

- För tumörbildning krävs att flera av barriärerna bryts som bilden visar vilket sker genom mutation eller inaktivering av molekyler
- Vanligt att både ONCO och suppressorer är påverkade
- Ringen behöver inte ske i ordning (men flera aktiveras), vid myelom behöver exempelvis inte vad ske?
Angiogenes

Fler Hallmarks
- Den växande klonen/tumören behöver ev också
- Genomisk instabilitet (DNA-reparationsgenerna t ex)
- Inflammatoriska processer (kan gynna del av tumörprogressionen)
2/4, vilka två har vi glömt?
- Förändra cellmetabolism så den gynnar tillväxt
- Inaktivering av immunförsvaret för att skydda tumörcellen

Förändring av cellmetabolism (Warburg-effekten) (Hallmark)
- Glukos metaboliseras och pyruvat bildas som går till mitokondrie och oxidativ fosforylering vilket kräver närvaro av syre
- Frånvaro av syre ger pyruvat som omvandlas till laktat (anaerob glykolys)
Vad innebär Warburg-effekten?
- Att prolifererande vävnad (embryotillväxt/tumörtillväxt) huvudsakligen producerar laktat även om tillgång på O2 finns
- Förklaring
- Mängden ATP är inte begränsande faktor i början av tumörutveckling
- Den använder glukosen vid glykolysen till, metabola intermediärer till lipidsyntes, nukleotidsynts och aminosyrasyntes, används som byggnadsblock för den nya cellen, detta behov är alltså viktigare än mängden ATP
- Laktatet som bildas går ut i hepatocyten där det ombildas till pyruvat och ombildas genom Coricykeln till glukos som kan återgå och tjänstgöra som näringskälla till tumören och fler intermediärer

- Mutationer av gener kan störa metabolismen i cellcykeln
- Isocitratdehydrogenas ombildar isocitrat till alfa-ketoglutarat vilket också är viktigt för att driva hydroxylering av HIF-1-alfa (håller dessa låga)
- Vad kan hända vid mutation av IDH1?
- Vid mutation i genen för isocitratdehydrogenas produceras istället hydroxiglutarat som stimulerar tumörbildning eftersom det inte längre sker hydroxilering av HIF-1-alfa
- I DNA finns metyleringar av cytosiner som blockerar transkription
- Alfa-ketoglutarat är kofaktor för att ta bort metyleringar på DNA
- Vid muterat IDH ges alltså inte kofaktorn alfa-ketoglutarat vilket ger ökad DNA-metylering (hypermetylering) vilket ger kompakt DNA där många funktioner stängs av (motverkar transkription), kallas för aberrant DNA metylering

Immunrespons (Hallmark)
- T-celler kan aktiveras att döda celler/organismer eller inaktiveras (finstämd)
- Cancerceller skapar molekyler som blockerar receptorer (på T-cellen) som i normalfallet aktiverar/inaktiverar T-celler
- Ge ett exempel på hur detta kan nyttjas kliniskt i behandling mot cancer med hjälp av antikroppar, specifikt PD1-hämmare?
- Behandling med antikroppar kan aktivera immunceller att attackera tumörcellerna
- PD-L1 på tumörcellen interagerar med PD-1 på T-cellen och hindrar immunförsvaret att aktiveras emot tumören.
- Genom att blockera PD-1 på T-cellen skyddas inte tumören längre på detta sätt.
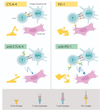
Immunrespons (Hallmark)
- Patientens T-celler plockas ut och utsätts för ett virus som uttrycker samma molekyl som tumörcellerna varpå T-cellen känner igen antigen på tumörcellens yta, fungerar väl på leukumi
- Vad kallas denna behandling för?
CAR T-cell terapi
Vilka två effekter har neovaskularisering?
- Perfussionen ger nutrienter och syre
- Nytt endotel ger stimulerar också till ytterligare tillväxt genom sekretion av IGFs och PDGF
- Det som startar angiogenesen är en övervikt mot angiogenespromotorer då de till en början oftast inte har denna egenskap vilket då gör att de stannar lokalt. Vilka celler initierar detta?
- Kan initieras av tumören själv (ex genom proteaser), inflammatoriska celler som makrofager, eller från stromala celler som svar på tumören
Vad frisätter proteaser från tumör eller stromala celler som svar på tumören?
FGF-2 som förvaras i ECM (viktig för proliferation och cellöverlevnad)
Brist på O2 stabiliserar HIF-1-alfa som då dimerar (HIF-1-beta) och aktiverar transkription av proangiogenetiska cytokiner från tumör, vilka är dessa och vad gör de?
VEGF och FGF som bägge stimulerar endotel till angiogenes i riktning mot tumör
Hur hör mutationer på p53 ihop med angiogenes?
Kan göra att dess antiangiogenesegenskaper går förlorade
- Transkriptionsfaktor för trombospondin (inhibitor av neovaskulisering)
- Repression av VEGF)
Detta utöver att cellens checkpoints kan påverkas
ONCO-gener som RAS, MAPK och MYC kan vid mutationer också uppregleras och ge?
VEGF
I vilka fyra steg sker invasion (ej lokal)?
Vad kan vi kalla detta med ett samlingsnamn?
Lösgöring, degradation, förändringar i kopplingar till ECM-komponenter, migration
Epitelial-mesenkymal-transition
Hur sker lösgöring från cell-cell-kontakt?
Genom inaktivering av E-cadherin
Hur sker degradering av ECM (basalmembran och interstitiellt matrix)?
Genom protealytiska enzymer från tumör/stromala celler som matrix metalloproteaser (MMPs), cathepsiner och kollagenaser
Utöver att bryta ner så utsöndrar metalloproteaser (MMPs), cathepsiner och kollagenaser något, vad?
Utsöndrar också tillväxtfaktorer vid nedbrytning av ECM och genererar kemotaxiska/angiogenes-komponenter)
Det sker alltså förändringar i kopplingar till ECM-komponenter, friska celler adhererar genom integriner till basalmembranet vilket ger G0 och vid förlust av dessa sker apoptos, hur lyckas tumörcellen undvika detta?
- detta är satt ur spel, dessutom är ECM efter klyvning av kollagen ex mer öppet för bindning av tumörreceptor och efterföljande migration
Hur sker migration av tumörceller?
Genom actin-cytoskelettrörelser mot ex kemotaktiska molekyler som IGF och klyvda kollagener/laminin, även parakrina faktorer. Detta är ett samspel mellan ECM, stromala celler (fibroblaster) och tumörcellen
Vilka tre steg sker vid metastasering?
Homing, adhesion och invasion av ny vävnad
Nämn ett par hinder för tumörcellen i cirkulationen
Shear stress, apoptos, immunförsvaret
Vad är signifikant för tumörcellen homing i cirkulationen?
Rör sig ofta i klumpar tillsammans eller med blodplättar (ibland emboli efter aktivering av koagelfaktorer)
Hur sker adhesion vid metastasering?
Genom integriner och laminreceptorer, specifikt exempel är CD44 i lymfoid vävnad
Ofta kan man se att invasion av ny vävnad hör ihop med primärtumörens placering och dess avgående blodcirkulation, de flesta metastaser ses vid första kapillärbädden efter (lunga och lever), men inte alltid vilket tyder?
Att adhesionsmolekyler och kemokinreceptorer med specificitet för vissa organ är inblandade. Att viss soil inte passande
Vad menas med att invasion av ny vävnad (metastasering) inte sker slumpmässigt?
Att den beror på egenskaper både hos de metastaserande cellerna och hos den invaderade vävnaden
Metastasering underlättas om tumörcellen producerar tillväxtfaktorer och angiogenesfaktorer. Den invaderade vävnaden är mer känslig om dessa celler också producerar tillväxtfaktorer och tillåter nykärlsbildning.
Immunrespons (Hallmark)
- T-celler kan aktiveras att döda celler/organismer eller inaktiveras (finstämd)
- Cancerceller skapar molekyler som blockerar receptorer (på T-cellen) som i normalfallet aktiverar/inaktiverar T-celler
Ge ett exempel på hur detta kan nyttjas kliniskt i behandling mot cancer med hjälp av antikroppar, specifikt CTLD-4-hämmare?
Vilka är riskerna?
- B7 känns först igen av CD28 men sen uppregleras CTLA-4 som då binder B7 och nedreglerar immunsvaret
- Men antikropp kan tillsättas som binder CTLA-4 och CD28 uppregleras igen vilket ger immunsvar
- Stor risk för autoimmunitet
- Tumörer med många mutationer svarar bäst
- Men antikropp kan tillsättas som binder CTLA-4 och CD28 uppregleras igen vilket ger immunsvar

Fler Hallmarks
Den växande klonen/tumören behöver ev också
- Förändra cellmetabolism så den gynnar tillväxt
- Inaktivering av immunförsvaret för att skydda tumörcellen
2/4, vilka två har vi glömt?
- Genomisk instabilitet (DNA-reparationsgenerna t ex)
- Inflammatoriska processer (kan gynna del av tumörprogressionen)
Nämn minst två transkriptionsfaktorer som är inblandade vid EMT (epitelial-mesenkymal-transition)
Snail, Slug, Twist, Zeb
