5 - ECZEMA, ATOPIC DERMATITIS, AND NONINFECTIOUS IMMUNODEFICIENCY DISORDERS Flashcards
What is eczema?
The word eczema seems to have originated in 543 ad and is derived from the Greek word ekzein, meaning to “to boil forth” or “to effervesce.” The term encompasses such disorders as dyshidrotic eczema and nummular eczema (NE), but at times is used synonymously for atopic dermatitis (atopic eczema). The acute stage generally presents as a red edematous plaque that may have grossly visible, small, grouped vesicles. Subacute lesions present as erythematous plaques with scale or crusting. Later, lesions may be covered by a drier scale or may become lichenified. In most eczematous reactions, severe pruritus is a prominent symptom. The degree of irritation at which itching begins (the itch threshold) is lowered by stress. Itching is often prominent at bedtime and usually results in insomnia. Heat and sweating may also provoke episodes of itching.
What is the hallmark of all eczematous eruptions?
Histologically, the hallmark of all eczematous eruptions is a serous exudate between cells of the epidermis (spongiosis), with an underlying dermal perivascular lymphoid infiltrate and exocytosis (lymphocytes present in overlying epidermis singly or in groups).
Spongiosis is generally out of proportion to the lymphoid cells in the epidermis. This is in contrast to mycosis fungoides, which demonstrates minimal spongiosis confined to the area immediately surrounding the lymphocytes.
In most eczematous processes, spongiosis is very prominent in the acute stage, where it is accompanied by minimal acanthosis or hyperkeratosis. Subacute spongiotic dermatitis demonstrates epidermal spongiosis with acanthosis and hyperkeratosis. Chronic lesions may have minimal accompanying spongiosis, but acute and chronic stages may overlap because episodes of eczematous dermatitis follow one another. Scale corresponds to foci of parakeratosis produced by the inflamed epidermis. A crust is composed of serous exudate, acute inflammatory cells, and keratin. Eczema, regardless of cause, will manifest similar histologic changes if allowed to persist chronically. These features are related to chronic rubbing or scratching and correspond clinically to lichen simplex chronicus or prurigo nodularis. Histologic features at this stage include compact hyperkeratosis, irregular acanthosis, and thickening of the collagen bundles in the papillary portion of the dermis. The dermal infiltrate at all stages is predominantly lymphoid, but an admixture of eosinophils may be noted. Neutrophils generally appear in secondarily infected lesions. Spongiosis with many intraepidermal eosinophils may be seen in the early spongiotic phase of pemphigoid, pemphigus, and incontinentia pigmenti, as well as some cases of allergic contact dermatitis.
chronic, inflammatory skin disease characterized by pruritus and a chronic course of exacerbations and remissions
Atopic dermatitis (AD)
It is associated with other atopic conditions, including food allergies, asthma, allergic rhinoconjunctivitis, eosinophilic esophagitis, and eosinophilic gastroenteritis. Because AD usually precedes the appearance of these other atopic conditions, it has been proposed that AD is the first step in an “atopic march” whereby sensitization to allergens through the skin may lead to allergic responses in the airways or digestive tract. Although this sequence of atopic conditions does occur in many children, whether the AD is causal in the development of the other manifestations of atopy is unproved but plausible. For this reason, early and effective treatment of AD is encouraged in an effort to prevent other atopic conditions. The genetic defect(s) predisposing at-risk individuals to the development of AD is the same for asthma and allergic rhinoconjunctivitis, and thus it has been difficult to prove that AD is causal in the development of other atopic conditions.
Epidemiology of Atopic dermatitis
The prevalence of AD, asthma, and allergic rhinoconjunctivitis increased dramatically in the last half of the 20th century, becoming a major health problem in many countries. The increase began first in the most developed nations, and as the standard of living has increased worldwide, so has the prevalence of AD. Rates of AD are about 30% in the most developed nations and exceed 10% in many countries, resulting in a worldwide cumulative prevalence of 20% In the most developed nations, the rates of AD plateaued in the 1990s, whereas developing nations have rates that continue to increase. Factors associated with high rates of AD are high latitude (perhaps associated with low levels of annual sun exposure) and lower mean annual temperature. A role for exposure to allergens thought to “trigger” AD is not supported by epidemiologic studies. Iceland has a very high rate of AD (27%) yet has no dust mites, few trees, and low pet ownership. However, children in Iceland often have positive skin prick tests to environmental allergens (24%). This questions the value of such tests in predicting causal environmental allergens in AD. Girls are slightly more likely to develop AD. In the United States an increased risk of AD during the first 6 months of life is noted in infants with African and Asian race/ethnicity, male gender, greater gestational age at birth, and a family history of atopy, particularly a maternal history of eczema. Other factors that increase the risk for the development of AD early in childhood include consumption of a Western diet, birth order (first children at greater risk), and delivery by cesarean section, all of which alter the intestinal microbiome. Exposure to antibiotics prenatally during the first or second and third trimesters also increases risk of AD. Therefore biodiversity in the gut microbiome seems to be protective. Gut colonization with Clostridium cluster I is associated with development of AD. Dog ownership before age 1 year decreases the risk of developing AD by age 4, but cat ownership has no effect. The hygiene hypothesis suggests that being raised on a farm lowers the risk of AD whereas living in modernized, cleaner indoor environments leads to a higher risk of AD.
About 50% of cases of AD appear in the first year of life, the vast majority within the first 5 years of life, and the remaining cases of “adult” AD usually before age 30. Atopy is now so common in the population that most individuals have a family history of atopy. Elevated immunoglobulin E (IgE) levels are not diagnostic of atopic disease in the adult. Therefore elevated IgE and a family history of “atopy” in an adult with new-onset dermatitis should not be used to confirm the diagnosis of adult AD Adult AD should only be considered when the dermatitis has a characteristic distribution and when other significant diagnoses, such as allergic contact dermatitis, photodermatitis, and cutaneous T-cell lymphoma, have been excluded. Rather, a dermatologist should infrequently make the diagnosis of adult “atopic dermatitis” for a dermatitis appearing for the first time after age 30.
Genetic Basis and Pathogenesis of atopic dermatitis
Eighty percent of identical twins show concordance for AD. A child is at increased risk of developing AD if either parent is affected. More than one quarter of offspring of atopic mothers develop AD in the first 3 months of life. If one parent is atopic, more than half the children will develop allergic symptoms by age 2. This rate rises to 79% if both parents are atopic. All of these findings strongly suggested a genetic cause for AD. Filaggrin is a protein encoded by the gene FLG, that resides in the epidermal differentiation complex (EDC) on chromosome 1q21. Filaggrin is processed by caspase 14 during terminal keratinocyte differentiation into highly hydroscopic pyrrolidone carboxylic acid and urocanic acid, collectively known as the “natural moisturizing factor” (NMF). Null mutations in FLG lead to reduction in NMF, which probably contributes to the xerosis that is almost universal in AD. Transepidermal water loss (TEWL) is increased. This may be caused by subclinical dermatitis, but also by abnormal delivery of lamellar body epidermal lipids (especially ceramide) to the interstices of the terminally differentiated keratinocytes. The resulting defective lipid bilayers retain water poorly, leading to increased TEWL and clinical xerosis. Ichthyosis vulgaris is caused by mutations in the FLG gene and is frequently associated with AD. Four FLG mutations have an estimated combined allelic frequency of 7%–10% in individuals of European descent. Different FLG gene mutations are associated with AD in other ethnicities although the rates do not necessarily match AD prevalence, demonstrating that the disease is multifactorial. Filaggrin 2 (FLG2), also in the EDC and w th similar function to FLG, is associated with persistent AD in African Americans. Inheriting one null FLG mutation slightly ncreases one’s risk of developing AD, and inheriting two mutations, either as a homozygote or a compound heterozygote, dramatically increases one’s risk. Between 42% and 79% of persons with one or more FLG null mutations will develop AD. However, 40% of carriers with FLG null mutations never have AD. FLG mutations are associated with AD that presents early in life, tends to persist into childhood and adulthood, and is associated with wheezing in infancy and with asthma. FLG mutations are also associated with allergic rhinitis and keratosis pilaris, independent of AD. Hyperlinear palms are strongly associated with FLG mutations, with a 71% positive predictive value (PPV) for marked palmar hyperlinearity. LAMA3 gene mutations, encoding the alpha chain of laminin 5, may also predispose to AD. In murine models, decreased Claudin 3 expression can lead to leakage of sweat in the superficial dermis contributing to impaired sweating in atopic dermatitis.
Not all cases of AD are associated with FLG mutations. AD patients often demonstrate immunologic features consistent with a T-helper 2 (Th2) phenotype, with elevated IgE, eosinophils on skin biopsy, and positive skin tests and radioallergosorbent test (RAST). The cytokines, especially interleukin-4 (IL-4) and interleukin-13 (IL-13), that are released due to the Th2 immune response play an important role in AD by increasing inflammatory cell infiltration and stimulating the inflammatory feedback loop. Basophils and innate type 2 lymphoid cells can also secrete IL-4 and IL-13. Thymic stromal lymphopoietin (TSLP) is an important interleukin-7 (IL-7)–like cytokine that, through its interaction with Th2 cells, basophils, mast cells, and dendritic cells, promotes the secretion and production of Th2 cytokines and the development of inflammatory Th2 CD4+ T cells (through production of OL40L). TSLP is produced by keratinocytes and is found in high levels in AD skin lesions. In addition, interleukin-31 (IL-31) is produced by Th2 and Th22 cells. IL-31 binds directly to nerves leading to itching and also downregulates expression of filaggrin. The JAKSTAT pathway is also critical in causing overactivation of the TH2 response and itch. Interleukin-17 (IL-17) and interleukin-22 (IL-22), which are released by Th17 cells, are also elevated in AD. Thus AD appears to represent a disorder characterized by a barrier defect that activates a specific Th2 response leading to a cycle of inflammation mediated by various inflammatory signals. The cytokines produced then worsen the already defective barrier. This leads to a vicious cycle of barrier failure and progressive inflammation, producing a chronic, relapsing, pruritic disorder, and explains why moisturization alone is not enough once the inflammatory cascade has started.
Preventionof AD in High-Risk Children
Extensive studies have been undertaken to determine whether it is possible to prevent the development of AD in children at high risk—those with parents or siblings with atopy. The most promising studies now repeated multiple times show that early moisturization, starting before 3 weeks of life, with thick emollient, may prevent AD in children at high risk of developing AD. Twice daily moisturization with various emollients such as petrolatum, sunflower seed oil, and others leads to an approximately 50% reduction in the expected rate of AD development in the first 6 months of life. This supports the idea that breaks in the skin barrier are an essential step in developing AD.
Switching to soy formula does not appear to reduce the risk of developing AD. Prolonged exclusive breastfeeding beyond 3–4 months of age is not protective for the development of AD. Extensively hydrolyzed casein formulas may be used as a supplement or substitute for breast milk during the first 4 months of life. Maternal allergen avoidance during pregnancy does not reduce the risk of AD in the offspring. Probiotic administration during and after pregnancy has been shown to decrease AD incidence by 14% based on data from a meta-analysis However, the type of probiotic to use, exactly when to start, and the safety during pregnancy are not fully elucidated. House dust mite (HDM) avoidance does not reduce AD, even in sensitized individuals, and high levels of HDMs in the environment in early life reduce AD risk.
Role of Food allergy in AD
The role of food allergy in AD is complicated, and the purported role of foods in AD has changed in recent years. Approximately 35% of children with moderate to severe AD have food allergies. However, 85% of children with AD will have elevated IgE to food or inhalant allergens, making a diagnosis of food allergy with serum or prick tests alone challenging due to the high false-positive rate of testing. Before food allergy testing is undertaken, treatment of the AD should be optimized. Parents are often seeking a “cause” for the child’s AD, when in fact it could be controlled with appropriate topical measures. Food restriction diets can be difficult and could put the child at risk for malnourishment and possibly development of allergy and should never be pursued without the oversight of an allergist and nutritionist to ensure proper nutrition. Food allergy should be pursued only in younger children with moderate to severe AD in whom standard treatments have failed. These children should also have a history of possible triggering of AD by specific food exposures. Testing, if performed, should be targeted foods to which the child is likely to be exposed, but generally wheat, egg, soy, cow’s milk, and peanut testing has been the most relevant to the AD. Double-blind placebo-controlled food challenges are
the “gold standard” for diagnosing food allergy. Skin prick tests have a high negative predictive value (>95%) but a PPV of only 0%–65%. For example, more than 8% of the U.S. population has a positive prick test to peanut, but only 0.4% are actually clinically allergic. Possible food allergy detected by testing should be confirmed by clinical history A positive RAST or skin prick test for a food that the child rarely or never ingests is probably not causally relevant to the child’s AD. Higher serum IgE levels and larger wheal sizes (>8–10 mm) are associated with greater likelihood of reacting to these foods when challenged. About 90% of food allergy is caused by a limited number of foods, as follows:
- Infants: cow’s milk, egg, soybean, wheat
- Children (2–10 years): cow’s milk, egg, peanut, tree nuts, fish, crustacean shellfish, sesame, kiwi fruit
- Older children: peanut, tree nuts, fish, shellfish, sesame, pollenassociated foods
Breastfeeding mothers must avoid the incriminated foods if their infant has been diagnosed with a food allergy.
There has been a rapid rise in peanut allergy in the United States. Recommendations to limit peanut exposure in childhood has been revised. A large randomized placebo control trial showed that early exposure in children at high risk for peanut allergy (due to strong family history of atopy) decreased the rate of development of peanut allergy by 86%. Therefore the revised guidelines that were developed in conjunction with dermatologists and allergists for peanut exposure are based on AD in the child. Of course a whole peanut should never be given to an infant due to choking hazard; therefore this was done with smooth peanut butter or the peanut snack Bamba (trademark).
1 Children with severe AD or egg allergy should be considered for peanut testing before exposure to peanuts around 4–6 months of age.
- Mild to moderate AD: Introduce peanuts around 6 months.
- No AD or food allergy: Introduce peanuts in accordance with family and cultural preferences.
Role of aeroallergens in AD
It is debated how much of a role aeroallergens play in the pathophysiology of AD. Although early exposure to allergens may lessen the incidence of AD, exposure to dust mites, animal allergens, mold, pollen, tree allergens, and other airborne allergens in those who are allergic may exacerbate AD. Testing for IgE responses to aeroallergens may not be predictive of their effect on patients with AD. In patients who note worsening around specific allergens, they should avoid these. Common allergens to avoid if possible include tobacco smoke, pollen, house dust mites, cats, and dogs (if allergic) although early exposure to dogs may prevent AD. Reduction of dust mite exposure can be achieved through vacuuming with a filtered vacuum, using dust mite covers on mattresses, avoidance of stuffed animals, and wall to-wall carpeting if feasible.
3 stages of AD
AD can be divided into three stages:
- infantile AD, occurring from 2 months to 2 years of age;
- childhood AD, from 2–10 years; and
- adolescent/adult AD.
What are the clinical manifestations of AD?
In all stages, pruritus is the hallmark. Itching often precedes the appearance of lesions, thus the concept that AD is “the itch that rashes.” Useful diagnostic criteria include those of Hannifin and Rajka, the UK Working Party, and the American Academy of Dermatology’s Consensus Conference on Pediatric Atopic Dermatitis (Boxes 5 1 and 5.2). These criteria have specificity at or above 90% but have much lower sensitivities (40%–100%). Therefore these criteria are useful for enrolling patients in studies and ensuring that they have AD, but less practical in diagnosing a specific patient with AD.
Hannifin and Rajka Criteria for Atopic Dermatitis

Modified Criteria for Children With Atopic Dermatitis

Infantile Atopic Dermatitis

Fifty percent or more of AD cases present in the first year of life, but usually not until after 2 months. Widespread dermatitis in children under 2 months may be from an irritant, ichthyosis, or a hallmark of severe immunodeficiency. AD in infancy usually begins as erythema and scaling of the cheeks (Fig. 5.1). The eruption may extend to the scalp, neck, forehead, wrists, extensor extremities, and buttocks. There can be overlap with seborrheic dermatitis on the scalp and in the folds, but papular or nodular involvement in the ax llae and inguinal folds is more typical of scabies infestation. Children with AD who have FLG gene mutations specifically have more cheek and extensor arm/hand involvement. There may be significant exudate; secondary effects from scratching, rubbing, and infection include crusts, infiltra ion, and pustules, respectively. Occlusion of saliva due to teething, extensive breastfeeding, and drooling may cause the cheeks and upper chest to flare and thick emollients can help prevent this. Breastfeeding should not be curtailed. The infiltrated plaques eventually take on a characteristic lichenified appearance The infantile pattern of AD usually disappears by the end of the second year of life.
Worsening of AD is often observed in infants after immunizations and viral infections likely due to activation of the immune system. Partial remission may occur during the summer, with relapse in winter. This may relate to the therapeutic effects of ultrav olet (UV) B light and humidity in many atopic patients, as well as the aggravation by wool and dry air in the winter. Extensive airborne environmental allergies may lead to worsening in the warmer months.
Childhood Atopic Dermatitis
Fig. 5.2 Flexural involvement in childhood atopic dermatitis.

During childhood, lesions tend to be less exudative. The classic locations are the antecubital and popliteal fossae (Fig. 5.2), flexor wrists, ankles, eyelids, face, and around the neck. Lesions are often lichenified, indurated plaques. These are intermingled with isolated, excoriated, 2–4 mm papules that are scattered more widely over the uncovered parts. Nummular morphology and involvement of the feet are more common in childhood AD.
Pruritus is a constant feature, and most of the cutaneous changes are secondary to it. Itching is paroxysmal. Scratching induces lichenification and may lead to secondary infection. A vicious cycle may be established, the itch-scratch cycle, as pruritus leads o scratching, and scratching causes secondary changes that in them cause itching. Instead of scratching causing pain, in the atopic patient the “pain” induced by scratching is perceived as itch and induces more scratching. The scratching impulse is beyond the control of the patient. Severe bouts of scratching occur during
sleep, leading to poor rest and chronic tiredness in atopic children. This can affect school performance. Parents often scold children who are scratching, and this leads to more anxiety and thus more scratching.
Severe AD involving a large percentage of the body surface area BSA) can be associated with growth retardation (Fig. 5.3). Restriction diets and steroid use may exacerbate growth impairment. Aggressive management of such children with phototherapy or systemic immunosuppressive agents may allow for rebound growth. Children with severe AD may also have substantial psychological disturbances. Parents should be questioned with regard to school performance and socialization. Although using light therapy and systemic immunosuppressive or immunomodulatory therapy in children can be daunting for patients and practitioners, the benefits to quality of life can dramatically outweigh the risks.
Fig. 5.3 Severe, widespread atopic dermatitis.

Atopic Dermatitis in Adolescents and Adults

Fig. 5.4 Prurigo-like papules in adult atopic dermatitis.
Most adolescents and adults with AD will give a history of childhood disease. AD will begin after age 18 years in only 6%–14% of patients diagnosed with AD. One exception is the patient who moves from a humid, tropical region to a more temperate area of higher latitude. This climatic change is often associated with the appearance of AD. In older patients, AD may occur as localized erythematous, scaly, papular, exudative, or lichenified plaques. In adolescents, the eruption often involves the classic antecubital and popliteal fossae, front and sides of the neck, forehead, and area around the eyes. In older adults, the distribution is generally less characteristic, and localized dermatitis may be the predominant feature, especially hand, nipple, or eyelid eczema. At times, the eruption may generalize, with accentuation in the flexures. The skin generally is dry and somewhat erythematous. Lichenification and prurigo-like papules are common (Fig. 5.4). Papular lesions tend to be dry, slightly elevated, and flat topped. They are almost always excoriated and often coalesce to form plaques. Staphylococcal colonization is common. In darker-skinned patients, the lesions are often hyperpigmented, frequently with focal hypopigmented areas related to healed excoriations.
Itching usually occurs in crises or paroxysms. Adults frequently complain that flares of AD are triggered by acute emotional upsets. Stress, anxiety, and depression reduce the threshold at which itch is perceived and result in damage to the epidermal permeability barrier, further exacerbating AD. Atopic persons may sweat poorly and may complain of severe pruritus related to heat or exercise. Physical conditioning and liberal use of emollients improve this component, and atopic patients can participate in competitive sports.
Even in patients with AD in adolescence or early adulthood, improvement usually occurs over time, and dermatitis is uncommon after middle life. In general, these patients retain mild stigmata of the disease, such as dry skin, easy skin irritation, and itching in response to heat and perspiration. They remain susceptible to a flare of their disease when exposed to a specific allergen or environmental situation. Photosensitivity develops in approximately 3% of AD patients and may manifest as either a polymorphous light eruption–type reaction or simply exacerbation of the AD by UV exposure. The average age for photosensitive AD is the middle to late thirties. Human immunodeficiency virus (HIV) infection can also serve as a trigger, and new-onset AD in an at-risk adult should lead to counseling and testing for HIV if warranted.
The hands, including the wrists, are frequently involved in adults, and hand dermatitis is a common problem for adults with a history of AD. It is common for irritant or atopic hand dermatitis to appear in young women after the birth of a child, when increased exposure to soaps and water triggers their disease. There is a new trend in children who can buy kits or mix their own various household liquids such as glue, borax, contact solution, baking soda and others to make “slime” This can lead to irritant dermatitis or worsening of hand atopic dermatitis. Wet work is a major factor in hand eczema in general, including those patients with AD.
Fig. 5.5 Atopic hand dermatitis.

Atopic hand dermatitis can affect both the dorsal and the palmar surface (Fig. 5.5). Keratosis punctata of the creases, a disorder seen almost exclusively in black persons, is also more common in atopic patients. Patients with AD have frequent exposure to preservatives and other potential allergens in the creams and lotions that are continually applied to their skin. Contact allergy may manifest as chronic hand eczema. Patch testing can help differentiate an allergic contact dermatitis.
Fig. 5.6 Periocular atopic dermatitis.

Eyelids are often involved (Fig. 5.6). In general, the involvement is bilateral and the condition flares with cold weather. As in hand dermatitis, irritants and allergic contact allergens must be excluded by a careful history and patch testing.
linear transverse fold just below the edge of the lower eyelids
Dennie-Morgan fold - indicative of the atopic diathesis, although it may be seen with any chronic dermatitis of the lower lids
In atopic patients with eyelid dermatitis, increased folds and darkening under the eyes is common.

When there is extensive facial involvement, the nose is still typically spared. This is called the ______
“headlight sign.”
The axillary vault and inguinal folds are also typically spared likely due to high humidity in these areas.

A prominent nasal crease may also be noted due to chronic upward wiping of the nose when there is rhinitis secondary to seasonal allergies. This is called the _____
“nasal salute.”
Other manifestations of AD
The less involved skin of atopic patients is frequently dry and slightly erythematous and may be scaly. Histologically, the apparently normal skin of atopic patients is frequently inflamed subclinically. The dry, scaling skin of AD may represent low-grade dermatitis. Pityriasis alba is a form of subclinical dermatitis, frequently atopic in origin. It presents as poorly marginated, hypopigmented, slightly scaly patches on the cheeks, upper arms, and trunk, typically in children and young adults with types III to V skin. It usually responds to emollients and mild topical steroids, preferably in an ointment base.
consists of horny follicular lesions of the outer aspects of the upper arms, legs, cheeks, and buttocks and is often associated with AD, AD and occurs in patients with filagrin mutations.

Keratosis pilaris (KP)
The keratotic papules on the face may be on a red background, a variant of KP called keratosis pilaris rubra faceii. KP is often refractory to treatment. Moisturizers alone are only partially beneficial. Some patients will respond to topical lactic acid, urea, or retinoids but they can easily irritate the skin of atopic patients and should be avoided in young children. If older patients desire, treatment should begin with applications only once or twice a week. KP must be differentiated from follicular eczema which tends to affect the trunk and is often more prominent in patients with skin ypes III-VI.
Thinning of the lateral eyebrows is sometimes present in AD

Hertoghe sign
This apparently occurs from chronic rubbing caused by pruritus and subclinical dermatitis. Hyperkeratos s and hyperpigmentation, which produce a “dirty neck” appearance, are also common in AD patients.
blanching of the skin at the site of stroking or scratching
White dermatographism
Chronic exposure to the vasoconstrictive effects of topical and oral steroids may lead to erythroderma due to vasodilation when steroids are tapered. This can lead to dysesthesias and belies the importance of using maintenance therapies that limit steroid exposure or lessen strength.
Atopic patients are at increased risk of developing various forms of urticaria, including contact urticaria. Episodes of contact urticaria may be followed by typical eczematous lesions at the affected site because skin that is scratched may flare with AD.
Ophthalmologic Abnormalities in AD
Up to 10% of patients with AD develop cataracts, either anterior or posterior subcapsular. Posterior subcapsular cataracts in atopic individuals are indistinguishable from corticosteroid-induced cataracts. Development of cataracts is more common in patients with severe dermatitis. Keratoconus is an uncommon finding, occurring in approximately 1% of atopic patients. Contact lenses, keratoplasty, and intraocular lenses may be required to treat this condition. Environmental allergies may also lead to allergic rhinoconjunctivitis.
Susceptibility to Infection of patients with AD
Fig. 5.7 Recurrent herpes simplex in atopic dermatitis.

Chronic eczematous lesions are often colonized with Staphylococcus aureus. Staphylococcus epidermitis may actually be protective as it is predominates in patients with fewer flares. In addition, the apparently normal nonlesional skin of atopic patients may also be colonized by S. aureus. The finding of increasing numbers of pathogenic staphylococci on the skin of a patient with AD is frequently associated with weeping and crusting of skin lesions, retroauricular and infraauricular and perinasal fissures, folliculitis, and adenopathy. In a flaring atopic patient, the possibility of
secondary infection must be considered. IgE antibodies directed against Staphylococcus and its toxins have been documented in some atopic individuals. Staphylococcal production of superantigens is another possible mechanism for staphylococcal flares of disease. Treatment of lesions of AD with topical steroids is associated with reduced numbers of pathogenic bacteria on the surface, even if antibiotics are not used. Despite the frequent observation that the presence of staphylococcal infection of lesions of AD is associated with worsening of disease, it has not been proven that oral antibiotics make a long term difference in the course of AD. Nonetheless, treatment of the “infected” AD patient with oral antibiotics is a community standard of dermatologists worldwide.
With the widespread presence of antibiotic-resistant S. aureus, dermatologists have shifted from the chronic use of oral antibiotics in managing patients with frequent flares of AD associated with staphylococcal infection. Rather, dilute sodium hypochlorite (bleach) baths and reduction of nasal carriage have become the basis for preventing infection-triggered AD. The typical formula is one quarter of a US cup (approximately 60 milliliters) of plain, unscented, NOT concentrated, NOT splashproof bleach per 20 gallon bathtub of water. In patients with AD and frequent infections, chronic suppressive oral antibiotic therapy may stabilize the disease. Options include cephalosporins, trimethoprim-sulfamethoxazole (TMP-SMX), clindamycin, and (in older patients) doxycycline. Identifying and treating S. aureus carriers in the family and pets may also be of benefit. An unusual complication of S. aureus infection in patients with AD is subungual infection, with osteomyelitis of the distal phalanx. In atopic patients with fever who appear very toxic, the possibility of streptococcal infection must be considered. These children may require hospital admission and intravenous antibiotics. Group A streptococcal superinfection of AD typically presents with a more vesiculopustular look that may mimic herpes simplex virus (HSV). Culture of the skin can differentiate and guide therapy typically with a penicillin- or cephalosporin-based antibiotic.
Patients with AD have increased susceptibility to viral superinfection, especially from HSV, varicella-zoster virus (VZV), enteroviruses (Coxsackie), vaccinia, and molluscum. Kaposi varicelliform infection is a term used to describe an acute vesicular eruption on top of AD and is usually caused by HSV, VZV, or enterovirus superinfection. Eczema herpeticum is seen most frequently in young children and is usually associated with HSV type 1 transmitted from a caretaker or sibling Once infected, there can be frequent flares of eczema herpeticum. Eczema herpeticum presents as the sudden appearance of vesicular, pustular, crusted, or eroded lesions concentrated in the areas of dermatitis (Fig. 5.7). The patients may have a fever and appear toxic. The lesions may continue to spread, and mos of the skin surface may become involved Secondary staphylococcal or streptococcal infection is common and local edema and regional adenopathy frequently occur. If lesions of eczema herpeticum occur on or around the eyelids, ophthalmologic evaluation is recommended. The severity of eczema herpeticum is quite variable, but most cases require sys emic antiviral therapy and an antistaphylococcal antibiotic. Delayed administration of acyclovir in hospitalized patients is associated with prolonged hospital stay. Genetic variants in TSLP and interferon regulatory factor 2 (IFR-2) in addition to DOCK 8 mutations are associated with AD and eczema herpeticum.
Recently a more virulent form of “hand foot and mouth” caused by enterovirus A6 instead of the typical A16 (also termed Coxackie A16) has been recognized and labeled “eczema coxsackium ” Superinfection with the A6 strain often leads to very widespread papules and vesicles with low-grade fever. There will typically be at least a few lesions on the palms and soles and in the mouth but there is not the typical predominance in these areas compared with areas of AD. Therapy is supportive.
Vaccination against smallpox is contraindicated in persons with AD, even when the dermatitis is in remission. Widespread and even fatal vaccinia can occur in patients with an atopic diathesis.
Atopic individuals may also develop extensive flat warts or molluscum contagiosum (MC). If the infection is extremely exuberant, an immunodeficiency such as DOCK-8 mutation should be ruled out. A common presentation of MC in children is a new patch of AD in an unusual area such as an axilla due to MC causing an immune response leading to a flare of AD. Chemical therapies such as salicylic acid and cantharidin may cause severe irritation within lesions of AD. Destruction with curettage (for molluscum), cryosurgery, or electrosurgery may be required to clear the lesions.
Give diferential diagnosis for AD
Typical AD in infancy and childhood is straightforward because of its characteristic morphology; predilection for symmetric involvement of the face, neck, and antecubital and popliteal fossae; and association with food allergy, asthma, and allergic rhinoconjunctivitis. Dermatoses that may resemble AD include seborrheic dermatitis (especially in infants), irritant or allergic contact dermatitis, nummular dermatitis, photodermatitis, scabies, and cases of psoriasis with an eczematous morphology. Certain immunodeficiency syndromes (see later discussion) may exhibit a dermatitis remarkably similar or identical to AD. In older patients with new onset dermatitis, cutaneous T cell lymphoma and allergic contact dermatitis should be considered.
Histopathologic findings of AD
The histology of AD varies with the stage of the lesion, with many of the changes induced by scratching. Hyperkeratosis, acanthosis, and excoriation are common
Staphylococcal colonization may be noted histologically. Although eosinophils may not be seen in the dermal infiltrate, staining for eosinophil major basic protein (MBP) reveals deposition in many cases.
Education and support measures for AD
Parental and patient education is of critical importance in the management of AD. In the busy clinic setting, dermatologists frequently have insufficient time to educate patients adequately regarding the multiple factors that are important in managing AD. Educational formats that have proved effective have been immediate nursing education on the correct use of medications, weekly evening educational sessions, and multidisciplinary day treatment venues. There are also now multiple online resources from major academic centers, professional societies, and associations that can be helpful. In all cases, “written action plans” outlining a “stepwise approach” have been important for parent/patient education. In addition, patients with chronic disease often become disenchanted with medical therapies or simply “burn out” from having o spend significant amounts of time managing their skin disease. The psychological support that can be incorporated into educational sessions can help motivate parents/patients and keep them engaged in the treatment plan. Having a child with AD is extremely stressful and generates significant stress within the family. Sleep is lost by both the patient and the parents. Supportive educational techniques and sometimes professional behavioral health consultation can help the family cope with this burden. In addition, the dermatologist must consider the complexity and time commitment of any prescribed regimen and ensure that the parents/patient understand and are committed to undertaking the treatments proposed.
Barrier repair in AD
AD patients have an impaired epidermal barrier The cornerstone of treatment and prevention of AD lies in addressing this problem. Patients should moisturize daily, especially immediately after bathing, ideally while the skin is still damp within a few minutes of getting out of the water. This may be with petrolatum or a petrolatum-based product, an oil-based product, a dimethiconebased product, vegetable shortening, or a “barrier repair” moisturizer that contains the essential lipids of the epidermal barrier. These special barrier repair moisturizers have similar benefits in AD to low-potency topical steroids. They are easier to apply and, if available to the patient, may enhance compliance. Petrolatum and petrolatum-based moisturizers are most often recommended and are often the least expensive. However, men with significant body hair, AD patients triggered by heat, and the very rare patient with true allergic contact dermatitis to petrolatum may be unable to tolerate petrolatum-based agents. Moisturizers should ideally be fragrance and formaldehyde free because these are two of the most common allergens in patients with AD. The ratio and content of ceramides in the epidermis of patients with AD is abnormal. Moisturizers with ceramides added have been shown to provide some benefit beyond the actual emollient. Patients should be instructed on the barrier-damaging properties of soaps, hot water, and scrubbing. The pH of the skin that promotes normal epidermal function is slightly acidic (the acid mantle) so synthetic detergents that have a more acidic pH are preferred to more basic soaps that have a high concentration of surfactant. Detergent use should be restricted to the axilla, groin, face, soles, and scalp. Oil-based cleansers can be used to “wash” the skin without water. For flares of AD, the soak and smear technique (soak in tub, then seal in water with a heavy moisturizer or medicated ointments) or wet dressings (wet wraps) with topical steroids can be very effective. In dry climates, AD patients may note some benefit with humidifiers. α-Hydroxy acid–containing products (lactic acid, glycolic acid) can be irritating and can exacerbate inflamed AD. These products should only be used for the xerosis of AD when there is absolutely no inflammation or pruritus.
Atimicrobial therapy for AD
When the AD patient has evidence of infection, treatment with topical or systemic antibiotics may be appropriate. Rather than treating once an infection occurs, it appears that the key in AD is to reduce nasal staphylococcal carriage preemptively and to keep the skin decolonized from Staphylococcus. Dilute sodium hypochlorite (bleach) baths have rapidly become a mainstay in AD patients. Twice-weekly bathing in a tepid bath with one quarter of a US cup (approximately 60 ml) of standard household bleach (5–6%) diluted in 20 gallons of water dramatically improves AD on the trunk and extremities, but less so on the face. For a smaller baby bathtub this is typically 2.5 mL = 1 2 teaspoon per gallon of water. Many bleach manufacturers make splashless or double-concentrated bleach, which should be avoided. This treatment combines decolonization of the skin with hydration, addressing two of the major factors in worsening of AD. Dilute bleach baths have been shown to decrease Nuclear Factor (NF) kappa-beta and thus may serve a role as a primary antiinflammatory therapy as well. Adequate moisturizing after bathing is critical. Intranasal application of mupirocin is beneficial in reducing nasal carriage although resistance is emerging. In 80% of families, at least one parent is carrying the same staphylococcal strain as a colonized AD child. If the AD patient has recurrent infections, other carriers in the family and their pets are sought and treated aggressively. Recurrent infections, especially furunculosis, are a cardinal feature of children and adults with AD who have systemic immunologic abnormalities, especially hyper-IgE syndrome.
Environmental factors in AD
Stress, heat, sweating, and external irritants may precipitate an attack of itching and flare in the AD patient Wool garments should be avoided. Addressing these triggers may improve the AD. Itch nerves are more active at higher emperatures, so overheating should be avoided. Irritants and allergens in the numerous products that AD patients may use can lead to flares of AD. Patients should avoid products that contain common allergens and should be evaluated for allergic contact dermatitis if a topical agent is associated with worsening of their AD.
Antipruritic therapy for AD
The primary treatment for the pruritus of AD is to reduce the severity of the AD. Antihistamines are frequently used for the pruritus of AD but are mainly beneficial for their sedative properties. If the patient has environmental allergies that lead to itch, the scratched skin can flare with AD, so in these patients long-acting histamine-2 blockers such as cetirizine, loratadine, and fexofenadine may help by decreasing the symptoms of the environmental allergies. Sedating antihistamines can lose some of their sedative effect if used consistently, so ideally they are only used when needed. Diphenhydramine, hydroxyzine, and doxepin can all be efficacious. Nonsedating antihistamines do not appear to benefit the pruritus of AD in standard doses. In some patients, gabapentin, selective serotonin reuptake inhibitors (SSRIs), mirtazapine, and even opiate antagonists may reduce pruritus. Applying ice during intense bouts of itch may help to “break” an itch paroxysm. Moisturizing lotions containing menthol, phenol, or pramoxine can be used between steroid applications to moisturize and reduce local areas of severe itch. More widespread use of topical doxepin (Sinequan) is limited by systemic absorption and sedation.
most common class of medications, along with moisturizers, used for the treatment of AD
Topical corticosteroids
They are effective and economical. Ointments are preferred because they can serve a do ble purpose as an emollient, they do not burn when applied, and they typically have fewer ingredients leading to lower of a chance of allergic contact dermatitis
In infants, low-potency steroid ointments, are preferred. Regular application of emollients must be emphasized Once corticosteroid receptors are saturated, additional applications of a steroid preparation contribute nothing more than an emollient effect. In most body sites, once-daily application of a corticosteroid is almost as effective as more frequent applications, at lower cost and with less systemic absorption and likely increased compliance. In some areas, twice-daily applications may be beneficial, but more frequent applications are almost never of benefit. Steroid phobia is common in parents and patients with AD. Less frequent applications of lower-concentration agents or intermittent use of steroids 2–5 times weekly, with emphasis on moisturizing, address these concerns. It can also be useful to point out that the human body must produce endogenous steroids daily to function Application of topical corticosteroids under wet wraps or vinyl suit occlusion especially after soaking in a tub of water (soak and smear) can increase efficiency. For refractory areas, a stronger corticosteroid may be used. A more potent molecule is more appropriate than escalating concentrations of a weaker molecule because the effect of the latter plateaus rapidly as receptors become saturated, and the potent medication should be stopped when the flare subsides. Because AD is an inflammatory cascade that must be halted, it is important not to undertreat. Undertreatment leads to loss of faith on the part of the patient/parents and prolongs the suffering of the patient. For severe disease, using more potent topical steroids in short bursts of a few days to a week can help gain con rol of the disease. In refractory and relapsing AD, twice-weekly steroid application to the areas that commonly flare may reduce flares. Another maintenance method is to mix 1 part of hydrocortisone 2.5% ointment in with 1 to 4 parts of the patient’s preferred emollient 2–5 days per week, lessening the amount this is used as tolerated.
In older children and adults, medium-potency steroids are often used, except on the face, where milder steroids or calcineurin inhibitors are preferred. For thick plaques and lichen simplex chronicus–like lesions, extremely potent steroids may be necessary. Ointments are more effective because of their moisturizing properties and require no preservatives, reducing the likelihood of allergic contact dermatitis. If an atopic patient worsens or fails to improve after the use of topical steroids and moisturizers, the possibility of allergic contact dermatitis to a preservative or the corticosteroids must be considered. Contact allergy to the corticosteroid itself can occur. Corticosteroid allergy seldom manifests as acute worsening of the eczema Instead, it manifests as a flare of eczema whenever the corticosteroid is discontinued, even for a day. This may be difficult to differentiate from stubborn AD.
Although the potential for local and even systemic toxicity from corticosteroids is real, the steroid must be strong enough to control the pruritus and remove the inflammation. Even in small children, strong topical steroids may be necessary in weekly pulses to control severe flares. Monitoring of growth parameters should be carried out in infants and young children.
Ideally prevention of AD flares is with just emollients, but patients with more severe disease often need some consistent antiinflammatory medicines (similar to the paradigm used for asthma). Maintenance therapy with topical steroids as discussed earlier can be with twice-weekly application, mixing a low-potency steroid in with an emollient or with nonsteroidal antiinflammatory medications.
Topical Calcineurin Inhibitors for AD
Topical calcineurin inhibitors (TCIs) such as tacrolimus or pimecrolimus offer an alternative to topical steroids. Systemic absorption is generally not significant with either of these agents.
Although a 0.03% tacrolimus ointment is marketed for use in children, it is unclear whether it really offers any safety advantage over the 0.1% formulation. Patients may experience a warm or stinging feeling in the skin when the medicines are first started. This tends to resolve and tolerability is improved if the ointment is applied to “bone-dry” skin Patients experience less burning if eczematous patches are treated initially with a corticosteroid, with transition to a TCI after partial clearing. Improvement tends to be steady, with progressively smaller areas requiring treatment. TCIs are particularly useful on the eyelids and face, in areas prone to steroid atrophy, when steroid allergy is a consideration, or when systemic steroid absorption is a concern. Tacrolimus is more effective than pimecrolimus, with tacrolimus 0.1% ointment equivalent to triamcinolone acetonide 0.1%, and pimecrolimus equivalent to a class V or VI topical corticosteroid. There is a black box warning on calcineurin inhibitors in the United States, but a recent study that followed patients after marketing found no increase in malignancy in over 26,000 patient-years studied.
an antiinflammatory topical PDE4 inhibitor introduced in 2017 for AD in children over 2 years.
Crisaborole
It can be used on the face and skinfolds without concern for striae but may sting when applied. It may be most useful as a maintenance medication or for mild to moderate disease.
Tar for AD
Crude coal tar 1%–5% in white petrolatum or hydrophilic ointment USP, or liquor carbonis detergens (LCDs) 5%–20% in hydrophilic ointment USP, is sometimes helpful for an area of refractory AD. Tar preparations are especially beneficial when used for intensive treatment for adults in an inpatient or day care setting, especially in combination with UV phototherapy. Goeckerman therapy with tar and UVB in a day treatment setting will lead to improvement in more than 90% of patients with refractory AD, and prolonged remission can be induced.
Phototherapy for AD
If topical modalities fail to control AD, phototherapy is another step on the therapeutic ladder. Narrow-band (NB) UVB is highly effective and has replaced broadband UV for treating AD. When acutely inflamed, AD patients may tolerate UV poorly. Initial treatment with soak and smear topical steroids or a systemic immunosuppressive may cool off the skin enough to institute UV treatments. Patients with significant erythema must be introduced to UV at very low doses to avoid nonspecific irritancy and flaring of the AD. Often, the initial dose is much lower and the dose escalation much slower than in patients with psoriasis. In acute flares of AD, UVA I can be used. For patients unresponsive to NB UVB, photochemotherapy with psoralen plus UVA (PUVA) can be effective but the benefits must outweigh the risks. It requires less frequent treatments, and can be given either topically (soak/ bath PUVA) or systemically (oral PUVA).
an IL-4 receptor inhibitor that thus blocks the f tion of IL-4 and IL-13
Dupilumab
It is an injection given every 2 weeks after a loading dose and decreases pruritus and Eczema Area and Severity Index (EASI) scores. It is the first targeted systemic therapy for AD. The most common significant side effect is keratoconjunctivitis.
Systemic corticosteroids in AD
In general, systemic corticosteroids should be avoided due to concerns for reexacerbation when the steroids are stopped. Some will use systemic steroids to control acute exacerbations. In patients requiring systemic steroid therapy, short courses (≤3 weeks) are preferred. If repeated or prolonged courses of systemic corticosteroids are required to control the AD, phototherapy or a steroid-sparing systemic therapy should be considered. Chronic corticosteroid therapy for AD frequently results in significant steroid-induced side effects such as striae, infection, boney changes, and growth delay. Osteoporosis in women requires special consideration and should be addressed with a bisphosphonate early in the course of therapy when bone loss is greatest. Preventive strategies, such as calcium supplements, vitamin D supplementation, bisphosphonates, regular exercise, and smoking cessation, should be strongly encouraged. Dual-energy x-ray absorptiometry (DEXA) scans are recommended.
Cyclosporine for AD
Cyclosporine (cyclosporin A) is highly effective in the treatment of severe AD, but the response is rarely sustained after the drug is discontinued. It is very useful to gain rapid control of severe AD Cyclosporine has been shown to be safe and effective in both children and adults, although probably tolerated better in children. Potential long-term side effects, especially renal disease and hypertension, require careful monitoring, with attempts to transition the patient to a potentially less toxic agent if possible. The dose range is 3–5 mg/kg in children and 150–300 mg in adults, with a better and more rapid response at the higher end of the dose range. Cyclosporin should not be used long term due to risks of renal and other toxicity. Rebound flaring of AD is possible and can be significant after stopping cyclosporin A, and a plan should be in place for this eventuality.
Methotrexate for AD
Methotrexate has a long history of safe use in children for other diseases. Recently studies have been published demonstrating that its efficacy is similar to cyclosporine, although the onset to action is significantly slower and it is not FDA approved for this indication. Doses are similar to what is used in psoriasis (0.3–0.6 mg/kg given once weekly (up to 25 mg) in children and 10–25 mg weekly in adults. Patients on methotrexate should take folic acid as well. The time to relapse with methotrexate may be longer than with cyclosporine. Switching from oral to subcutaneous administration may increase the benefit.
Other Immunosuppressive Agents for AD
Several other immunosuppressive agents have demonstrated efficacy in patients with AD. They include axathioprine and mycophenolate mofetil. These agents do not appear to be as quick to work as cyclosporine. Patients with FLG mutations respond to azathioprine and methotrexate, but the response is slower than in those without FLG mutations. However, over time they may have a better safety profile, so patients requiring long-term immunosuppression may benefit from one of these agents. The dosing of azathioprine is guided by the serum thiopurine methyltransferase level. Mycophenolate mofetil (MMF) is generally well tolerated and, as with azathioprine, takes about 6 weeks to begin to reduce the AD. Unfortunately, the response of AD patients to MMF is variable, with 20%–40% not responding. If cyclosporin A is not to be used, the choice of steroid-sparing agent is personalized to the patient’s risk factors and tolerance of the medication. Hydroxyurea, as used for psoriasis in the past, can be effective in AD if other steroidsparing agents fail, as well as in the patient with liver disease.
Intravenous immune globulin (IVIG) has had some limited success in managing AD, but its high cost precludes it use, except when other reasonable therapeutic options have been exhausted. There are also reports of a dyshidrotic flare of dermatitis on the hands in patients on IVIG. Interferon-γ (IFN-γ) given by daily
injection has demonstrated efficacy in both children and adults with severe AD but is very rarely used. IFN γ is well tolerated but can cause flulike symptoms. Omalizumab can be considered in refractory cases, but only 20% of patients achieve a 50% or greater reduction of their AD. Infliximab has not been beneficial in AD. Ustekinumab has been effective in a few reports, because the inflammatory cascade triggering and maintaining AD does involve Th17 cells.
Traditional Chinese herb mixtures have shown efficacy in children and in animal models for AD. The active herbs appear to be ophiopogon tuber and Schisandra fruit. Chinese herbs are usually delivered as a brewed tea to be drunk daily. Their bitter taste makes them difficult to palate, and there is a risk of allergy from herbal products.
Management of acute flare of AD
Initially, the precipitating cause of the flare should be sought. Recent stressful events may be associated with flares. Secondary infection with S. aureus is one of the most common causes for flares and will manifest with pustules and honey-colored erosions. Less frequently, HSV or coxsackievirus may be involved. Pityriasis rosea may also cause AD to flare. The development of contact sensitivity to an applied medication or photosensitivity must be considered.
In the patient with an acute flare, treating triggers may lead to improvement (see earlier discussion). Soaking and smearing (wet wrapping) is recommended as first-line treatment. Often, 3–4 days of such intensive home therapy will break a severe flare. A short course of systemic corticosteroids may be of benefit as a last resort.
Fig. 5 8 Ear eczema.

Eczema of the ears or otitis externa may involve the helix, postauricular fold, and external auditory canal. By far the most frequently affected site is the external canal, where eczema is often a manifestation of seborrheic dermatitis or allergic contact dermatitis caused by topical medications, especially neomycin (Fig. 5.8), benzocaine, and preservatives or earphones. Secretions of the ear canal derive from the specialized apocrine and sebaceous glands, which form cerumen. Rubbing, wiping, scratching, and picking exacerbate the condition. Secondary bacterial colonization or infection is common. Infection is usually caused by staphylococci, streptococci, or Pseudomonas. Pseudomonas aeruginosa can result in malignant external otitis with ulceration and sepsis. Earlobe dermatitis in people with pierced ears is virtually pathognomonic for metal contact dermatitis (especially nickel).
Treatment should be directed at removal of causative agents, such as topically applied allergens. First, examine the ear with an otoscope and be sure there is not a perforated tympanic membrane. If there is drainage from a perforated tympanic membrane, management should be in consultation with an otolaryngologist. This purulent fluid can be the cause of an ear eczema—infectious eczematoid dermatitis. If the tympanic membrane is intact, scales and cerumen should be removed by gentle lavage with an ear syringe. A topical steroid otic solution can be used for noninfectious causes. For very weepy lesions, aluminum acetate optic solution (e.g., Domeboro) may be drying and beneficial.
Eyelid Dermatitis
Eyelid dermatitis is most often related to AD or allergic contact dermatitis, or both (see Chapter 6). Allergic conjunctivitis in an atopic patient may lead to rubbing and scratching of the eyelid and result in secondary eyelid dermatitis. Seborrheic dermatitis,
psoriasis, and airborne dermatitis are other possible causes. Most patients with eyelid dermatitis are female. When an ocular medication contains an allergen, the allergen passes through the nasolacrimal duct, and dermatitis may also be noted below the nares in addition to the eyelids. Some cases of eyelid contact dermatitis are caused by substances transferred by the hands to the eyelids. If eyelid dermatitis occurs without associated AD, an allergen is detected in more than 50% of cases. More than 25% of patients with AD and eyelid dermatitis will also have allergic contact dermatitis contributing to the condition. Fragrances and balsam of Peru, metals (nickel and gold), paraphenylenediamine, quaternium 15, oleamidopropyl dimethylamine, thiuram (in rubber pads used to apply eyelid cosmetics), and tosylamide formaldehyde (in nail polish) are common allergens causing eyelid dermatitis. In medications, preservatives such as cocamidopropyl betaine and active agents such as phenylephrine hydrochloride, sodium cromoglycate, papain, and idoxuridine have all been implicated.
Eyel d dermatitis requires careful management, often in collaboration with an ophthalmologist. The most important aspect is to identify and eliminate any possible triggering allergens as noted previously. Patch testing for standard allergens, as well as the patient’s ocular medications, is highly recommended. Preservative-free eye medications should be used. The ophthalmologist should monitor the patient for conjunctival complications, measure the intraocular pressure, and monitor for the development of cataracts, especially in patients with AD who have an increased risk for cataracts. Initially, topical corticosteroids and petrolatumbased emollients are recommended. If the dermatitis is persistent, the patient may be transitioned to TCIs or crisaborole to reduce the long-term risk of ocular steroid complications. The TCIs are often not initially tolerated on inflamed eyelids due to the burning. If there is an associated allergic conjunctivitis, or in patients who fail treatment with topical medications applied to the eyelid, ocular instillation of cyclosporine ophthalmic emulsion (Restasis) can be beneficial. Cromolyn sodium ophthalmic drops may be used to stabilize mast cells in the eyelid and reduce pruritus. In balsam of Peru–allergic patients, a balsam elimination diet may benefit.
Breast Eczema (Nipple Eczema)
Fig. 5.9 Nummular eczema of the breast.

Eczema of the breasts usually affects the areolae and may extend onto the surrounding skin (Fig. 5.9). The area around the base of the nipple is usually spared, and the nipple itself is less frequently affected. The condition is more common in women but can be seen in infants as well. Usually, eczema of the nipples is of the moist type with oozing and crusting. Painful fissuring is frequently seen, espec ally in nursing mothers. AD is a frequent cause, and nipple eczema may be the sole manifestation of AD in adult women. It frequently presents during breastfeeding. The role of secondary infection with bacteria and Candida should be considered in breastfeeding women. Other causes of nipple eczema are allergic contact dermatitis and irritant dermatitis. Irritant dermatitis occurs from friction (jogger’s nipples), or from poorly fitting brassieres with seams in women with asymmetric and large breasts. In patients in whom eczema of the nipple or areola has persisted for more than 3 months, especially if it is unilateral, a biopsy is mandatory to rule out the possibility of Paget disease of the breast. Topical corticosteroids or TCIs are often effective in the treatment of non-Paget eczema of the breast. Nevoid hyperkeratosis of the nipples is a chronic condition that may mimic nipple eczema, but it is not responsive to corticosteroids.
Nipple eczema in the breastfeeding woman is a therapeutic challenge and can be from AD, ACD, irritant dermatitis, infection or food allergy. A lactation consultant or nurse may be helpful in managing these patients, because poor positioning during breastfeeding is a common cofactor in the development of nipple eczema. The dermatitis may appear in an atopic woman when her child begins to ingest solid foods rarely due to contact dermatitis to a
food. Allergic contact dermatitis may develop to topical protective creams (containing vitamin A and E, aloe, lanolin, chamomile, or preservatives). Staphylococcal superinfection may develop and can be identified by culture. Oral antibiotics are the preferred treatment for bacterial secondary infection.
Candidal infection of the areola may present as normal skin, erythema, or an acute or chronic eczema. The area of the areola immediately adjacent to the nipple tends to be involved, sometimes with fine hairline cracks. Associated conditions include oral thrush in the infant, antibiotic use, and a personal history of vaginal candidiasis in mother. Cultures are typically positive from the affected areola/nipple. Oral thrush in the infant in the setting of nipple eczema in the mother would warrant treatment of the mother and infant. Patients frequently complain of severe pain, especially with nursing. Analgesia may be required, and breastfeeding may need to be suspended for a period. Pumping and the use of a silicone nipple shield may be helpful. Therapy with topical or systemic antifungal agents may be required. Topical gentian violet 0.5%, applied once daily to the nipple for up to 1 week, may be helpful but should not be ingested by the infant. Guaiazulene (Azulon) is a dark-blue hydrocarbon used in Europe for nipple “cracks” with breastfeeding.
Hand Eczema

Hand eczema is a common and important skin condition. The genetic risk factors for the development of hand dermatitis are unknown but patients with AD are more susceptible. The risk for persistence of the hand eczema is doubled if there is associated eczema at other sites at presentation, if there is a childhood history of AD, and if the onset of the hand eczema was before age 20. Atopic patients with the FLG null mutation may have a specific form of hand dermatitis characterized by dorsal hand and finger dermatitis, volar wrist involvement, and hyperlinear palms, but limited palmar dermatitis. Hand eczema is the most common occupational skin condition, accounting for more than 80% of all occupational dermatitides. About 1 per 1000 workers are affected annually. Women are at increased risk, most of which is accounted for by a “spike” in the rate of hand eczema in the 20–29 age-group, when increased environmental exposures increase women’s risk (e.g., child care). As noted above, children with new hand dermatitis
should be asked about making or handling “slime” which is an often home-made play substance that can cause irritant or allergic contact dermatitis.
Chronic hand eczema, especially if severe, significantly reduces the patient’s quality of life and can be associated with symptoms of depression. Persons at high risk for hand eczema can be identified and counseled to avoid high-risk occupations such as those that involve consistent exposure to water or sensitizing chemicals such as hairdressers. If occupational hand eczema develops, some occupation-specific strategies can lead to improvement and prevent recurrence.
The evalua ion and management of hand eczema has been hampered by the lack of a uniform classification system and a dearth of controlled therapeutic trials. The diagnostic dilemma in hand dermatitis is in part related to two factors. The clinical appearance of the skin eruption on the palms and soles may be very similar, independent of the etiology. In addition, virtually all chronic hand dermatitis demonstrates a chronic dermatitis histologically, again independent of pathogenic cause. For instance, psoriasis on the palms and soles may show the same spongiosis as a dermatitis (Fig. 5.10). As a result, the proposed classification schemes rely on a combination of morphologic features, history of coexistent illnesses, occupational exposure, and results of patch testing.
What are the different types of eczema?
- Allergic contact dermatitis (with or without an additional irritant component)
- Irritant hand dermatitis
- Atopic hand eczema (with or without an additional irritant component) 4 Recurrent vesicular (or vesiculobullous) hand eczema
- Hyperkeratotic hand eczema
- Pulpitis (chronic fingertip dermatitis)
- Nummular dermatitis
How do you establish diagnosis of hand eczema?
A complete history, careful examination of the rest of the body surface, and frequently patch testing are essential in establishing a diagnosis. Patch testing is recommended in all patients with chronic hand eczema. Allergens in the environment, especially shower gels and shampoos, in the workplace, and in topical medications may be important in any patient. Patch tes ing must include broad screens of common allergens or allergic contact dermatitis may be missed.
The role of ingested nickel in the development of hand eczema in nickel-allergic patients is controversial. Some practitioners treat such patients with low-nickel diets and even disulfiram chelation with reported benefit. However, the risk of development of hand eczema in adulthood is independent of nickel allergy. Similarly, the role of low-balsam diets in the management of balsam of Peru–allergic patients with hand eczema is unclear.
How do you define wet work?
Wet work, defined as skin in liquids or gloves for more than 2 hours per day, or handwashing more than 20 times per day, is a strong risk factor for hand eczema.
High-risk occupations include those that entail wet work and those with exposure to potential allergens. “high-risk” occupations include bakers, hairdressers, dental surgery assistants, kitchen workers/cooks, butchers, health care workers, cleaners, physicians/dentists/veterinarians, and laboratory technicians. In about 5% of patients with hand eczema, especially if severe, it is associated with prolonged missed work, job change, and job loss. In health care workers, the impaired barrier poses a risk for infection by blood-borne pathogens.
Almost one third of baker’s apprentices develop hand dermatitis within 12 months of entering the profession Among hairdressers, the incidence approaches 50% after several years. Both irritant dermatitis and allergic contact dermatitis are important factors, with glyceryl monothioglycolate and ammonium persulfate being the most common allergens among hairdressers. Cement workers have a high rate of hand dermatitis related to contact allergy, alkalinity, and hygroscopic effects of cement. Dorsal hand dermatitis in a cement worker suggests contact allergy to chromate or cobalt. The addition of ferrous sulfate to cement has no effect on irritant dermatitis, but reduces the incidence of allergic chromate dermatitis by two thirds.
Among patients with occupational hand dermatitis, atopic patients are disproportionately represented. Hand dermatitis is frequently the initial or only adult manifestation of an atopic diathesis. The likelihood of developing hand eczema is greatest in patients with AD, more common if the AD was severe, but is s ill increased in incidence in patients with only respiratory atopy. One third to one half of patients with hand eczema have atopy. Atopic patients should receive career counseling in adolescence to consider avoidance of occupations that are likely to induce hand dermatitis.
Contact urticaria syndrome may present as immediate burning, itching, or swelling of the hands, but a chronic eczematous phase may also occur. Latex is an important cause of the syndrome, but raw meat, lettuce, garlic, onion, carrot, tomato, spinach, grapefruit, orange, radish, fig, parsnip, cheese, or any number of other foods may be implicated.
What are the primary lesions of dyshidrosis?
Vesiculobullous Hand Eczema (Pompholyx, Dyshidrosis).
Primary lesions of dyshidrosis are deep-seated multilocular vesicles resembling tapioca on the sides of the fingers (Fig. 5.11), palms, and soles.
Fig. 5 11 Pompholyx.
The eruption is symmetric and pruritic, with pruritus often preceding the eruption. Coalescence of smaller lesions may lead to bulla formation severe enough to prevent ambulation. Individual outbreaks resolve spontaneously over everal weeks. Acute pompholyx, also known as cheiropompholyx if it affects the hands, presents with severe, sudden outbreaks of intensely pruritic vesicles. Idiopathic acute vesicular hand dermatitis is not related to blockage of sweat ducts, although palmoplantar hyperhidrosis is common in these patients, and control of hyperhidrosis improves the eczema. Bullous tinea or an id reaction from a dermatophyte should be excluded, and patch testing should be considered to rule out allergic contact dermatitis.

Chronic Vesiculobullous Hand Eczema
In chronic cases, the lesions may be hyperkeratotic, scaling, and fissured, and the “dyshidrosiform” pattern may be recognized only during exacerbations. The pruritic 1–2 mm vesicles tend to be most pronounced at the sides of the fingers. In long-standing cases, the nails may become dystrophic. The distribution of the lesions is, as a rule, bilateral and roughly symmetric.
The eruption presents as hyperkeratotic, fissure-prone, erythematous areas of the middle or proximal palm. Vesicles are not seen The volar surfaces of the fingers may also be involved (Fig. 5.12).

Fig. 5.12 Hyperkeratotic hand dermatitis.
Plantar lesions occur in about 10% of patients. Males outnumber females by 2 : 1, and the patients are usually older adults. Histologically, the lesions show chronic spongiotic dermatitis. The most important differential diagnosis is psoriasis, and some of the patients with chronic hyperkeratotic hand dermatitis will ultimately prove to be psoriatic. The presence of sharply demarcated plaques, nail pitting, or occasional crops of pustules is an important clue to psoriatic hand involvement.
This hyperkeratotic and fissuring eczema affects primarily the fingertips and may extend to merge with eczema of the palm.
Pulpitis (Fingertip Hand Dermatitis).
Vesicles can occur. Involvement of the three fingers of the dominant hand suggests a contact dermatitis (irritant or allergic), whereas similar involvement of the nondominant hand suggests vegetables and other items related to food preparation that are held in this hand for cutting (e.g., garlic).
What is the management for hand dermatitis?
The hands are essential for work both in and out of the home. Treatment regimens must be practical and must allow patients to function as normally as possible. The efficacy of some of the treatments depends on the morphology of the eruption and the diagnostic classification (see previous discussion).
Protection. Vinyl gloves may be worn during wet work, especially when detergents are used. Although vinyl gloves protect against chemicals, they do not prevent exposure to heat through the glove or the macerating effect of sweat, which accumulates under the gloves. Wearing white cotton gloves under the vinyl gloves can prevent this. Vinyl gloves are also much less durable than rubber gloves. Rubber gloves may be used at home if patients do not exhibit allergy to rubber chemicals or latex. For rough work, such as gardening, wearing protective cloth or leather gloves is essential.
Barrier Repair. Moisturizing is a critical component of the management of hand dermatitis. Application of a protective moisturizing cream or ointment after each handwashing or water exposure is recommended. Creams require a preservative and have a higher risk of contact sensitivity and often burn when applied. Ointments tend to have few ingredients and do not generally require a preservative. At night, even during periods of remission, a heavy moisturizing ointment should be applied to the hands after soaking in water. If palmar dryness is present, occlusion of the moisturizer with a plastic bag or vinyl gloves is recommended. White petrolatum is inexpensive and nonsensitizing and remains a valuable agent in the treatment of hand dermatitis. Jars of moisturizers can be contaminated with S. aureus.
Topical Agents. Potent and ultrapotent topical corticosteroids are first-line pharmacologic therapy. Their efficacy is enhanced by presoaking and occlusion (soak and smear technique or wet dressings). A single application with occlusion at night is often more effective than multiple daytime applications. The treatment is continued until the hands are clear, and then either emollients are substituted or maintenance treatment with the topical steroid two or three times weekly is continued to prevent recurrence. In refractory cases, ultrapotent corticosteroids may be used for 2–3 weeks, then on weekends, with a milder corticosteroid applied during the week.
The TCIs may be of benefit in some mildly affected patients. Soaks with a tar bath oil or applications of 20% LCD or 2% crude coal tar in an ointment base may be of benefit, especially in patients with hyperkeratotic hand eczema. Bexarotene gel can be beneficial in up to 50% of patients with refractory hand eczema.
Phototherapy. Phototherapy in the form of high-dose UVA I, soak or cream PUVA, and oral PUVA can be effective but the risks of chronic photo damage and skin cancer must be discussed with patients.. Given the thickness of the palms, UVA irradiation should be delivered 30 minutes after soaking, as opposed to bath PUVA, which can be done immediately after bathing.
Grenz ray radiotherapy is rarely used due to long-term risk of malignancy.
Botulinum Toxin A. In patients with palmoplantar hyperhidrosis and associated hand eczema, treatment of the hyperhidrosis with intradermal injections of botulinum toxin A leads to both dramatic resolution of the sweating and clearing of the hand eczema. The hand eczema returns when the sweating returns.
Iontophoresis, which also reduces sweating, can similarly improve hand dermatitis. This illustrates the importance of wetness in the exacerbation of hand eczema.
Systemic Agents. The systemic agents used to treat severe chronic hand dermatitis are identical to those used for AD. Systemic steroids are recommended only to control acute exacerbations. For example, patients with infrequent but severe outbreaks of pompholyx may benefit from a few weeks of systemic steroids, starting at about 1 mg/kg/day. Patients with persistent, severe hand dermatitis should be considered for alternative, steroid-sparing therapy. Oral retinoids may have a place in the management of hand dermatitis. Alitretinoin (not available in the United States),
may lead to complete or near-complete clearance of chronic refractory hand eczema in about 50% of patients, especially those with hyperkeratotic hand eczema. The onset of response is delayed, with some patients achieving optimal benefit only after more than 6 months of treatment. Acitretin, may have similar benefit and of course should not be used in women of childbearing potential.
Workplace Modifications. The incidence of hand dermatitis in the workplace can be reduced by identifying major irritants and allergens, preventing exposure through engineering controls, substituting less irritating chemicals when possible, enforcing personal protection and glove use, and instituting organized worker education. Hand eczema classes have been documented to reduce the burden of occupational dermatitis. It is important to note that prevention of exposure to a weak but frequent irritant can have more profound effects than removal of a strong but infrequently contacted irritant.
Proper gloves are essential in industrial settings. Nitrile gloves are generally less permeable than latex gloves. Gloves of ethylene vinyl alcohol copolymer sandwiched with polyethylene are effective against epoxy resin, methyl methacrylate, and many other organic compounds. Latex and vinyl gloves offer little protection against acrylates. The 4H (4 hour) glove and nitrile are best in this setting. As hospitals transition to nonlatex gloves, it is important to note that even low-protein, powder-free latex gloves reduce self-reported skin problems among health workers.
Diaper (Napkin) Dermatitis
Diaper dermatitis has dramatically decreased as a result of highly absorbable disposable diapers. Nonetheless, dermatitis of the diaper area in infants remains a common cutaneous disorder. The highest prevalence occurs between 6 and 12 months of age. Diaper dermatitis is also seen in adults with urinary or fecal incontinence who wear diapers.
Irritant diaper dermatitis is the most common type of dermatitis and is an erythematous dermatitis due to skin contact with urine and feces that is usually limited to the convex exposed surfaces. The folds remain unaffected, in contrast to intertrigo, inverse psoriasis, and candidiasis, where the folds are frequently involved. In severe cases of irritant dermatitis, there may be superficial erosion or even ulceration (Jacquet erosive diaper dermatitis), violaceous plaques and nodules (granuloma gluteal infantum), or pseudoverrucous papules and nodules; these three entities are part of a disease spectrum and can simulate herpetic infections or genital warts The tip of the penis may become irritated and crusted, with the baby urinating frequently and spots of blood appearing on the diaper.
Excessive hydration with maceration of the skin is the primary causal factor in diaper dermatitis. The absence of diaper dermatitis in societies where children do not wear diapers clearly implicates the diaper environment as the cause of the eruption. Many parents will incorrectly switch to cloth diapers when diaper dermatitis occurs even though the superabsorbent modern diapers are much more effective at preventing diaper dermatitis by wicking urine and stool to some extent away from the skin. Moist skin is more easily abraded by friction of the diaper as the child moves. Wet skin is more permeable to irritants. Skin wetness also allows the growth of bacteria and yeast. Bacteria raise the local pH, increasing the activity of fecal lipases and proteases, which leads to more skin breakdown. Candida albicans is frequently a secondary invader and, when present, produces typical satellite erythematous lesions or pustules at the periphery as the dermatitis spreads. S. aureus and group A β-hemolytic streptococci can infect diaper dermatitis. Breastfeeding is associated with less frequent diaper dermatitis, and diarrhea is a risk factor.
Give diferential diagnosis of diaper dermatitis
The differential diagnosis of diaper dermatitis should include napkin psoriasis (Fig. 5.13), seborrheic dermatitis, AD, langerhans cell histiocytosis, tinea cruris, acrodermatitis enteropathica, aminoacidurias, biotin deficiency, and congenital syphilis. Allergic contact dermatitis is becoming more frequently recognized as a cause of dermatitis in the diaper area. Allergens include sorbitan sesequioleate, fragrances, disperse dye, cyclohexylthiopthalimide, and mercaptobenzothiazole (in rubber diaper covers). Given the skill of most pediatricians in the management of diaper dermatitis, dermatologists should think about these conditions in infants who have failed the standard interventions used by pediatricians. Refractory diaper dermatitis may require a biopsy to exclude some of these conditions.

Fig. 5.13 Napkin psoriasis.
Prevention and treatment of diaper dermatitis
Prevention is the best treatment. Diapers that contain superabsorbent gel have been proved effective in preventing diaper dermatitis in both neonates and infants. They work by absorbing the wetness away from the skin and by buffering the pH. Cloth diapers and regular disposable diapers are equal in their propensity to cause diaper dermatitis and are inferior to the superabsorbent gel diapers. The frequent changing of diapers is also critical: every 2 hours for newborns and every 3–4 hours for older infants. The renewed popularity of cloth and bamboo diapers as more natural and ecologic has led to a reemergence of severe diaper dermatitis in some European countries.
Protecting the skin of the diaper area is vital. Zinc oxide paste and petrolatum are both effective barriers, preventing the urine and stool from contacting the dermatitis. If simple improved hygiene and barrier therapy are not effective, the application of anticandidal agents in addition to a very-low-potency topical steroid for a few days can aid in healing
Circumostomy Eczema
Eczematization of the surrounding skin frequently occurs after an ileostomy or colostomy. It is estimated that 75% of ileostomy patients have some postoperative sensitivity as a result of the leakage of intestinal fluid onto unprotected skin. As the consistency of the intestinal secretion becomes viscous, the sensitization subsides Proprietary medications containing karaya powder have been helpful; 20% cholestyramine (an ion-exchange resin) in a petrolatum-based moisturizer and topical sucralfate as a powder or emollient are effective treatments. Absorbent silicone protective layers can be placed around the ostomy to provide protection against the tube rubbing and absorption of any leaking intestinal contents. Psoriasis may also appear at ostomy sites, especially in patients with inflammatory bowel disease (IBD) being treated with tumor necrosis factor (TNF) inhibitors who develop psoriasis as a complication. Topical treatment may be difficult because the appliance adheres poorly after the topical agents are applied. A topical corticosteroid spray may be used and will not interfere with appliance adherence. Contact dermatitis to the ostomy bag adhesive can be problematic, and even supposedly hypoallergenic ostomy bags may still trigger dermatitis in these patients.
refers to the spontaneous development of widespread dermatitis or dermatitis distant from a local inflammatory focus
Autoeczematization (id reaction)
The agent causing the local i matory focus is not the direct cause of the dermatitis at the distant sites. Autoeczematization most frequently presents as a generalized acute vesicular eruption with a prominent dyshidrosiform component on the hands. The most common associated condition is a chronic eczema of the legs, with or without ulceration. The “angry back” or “excited skin” syndrome observed with strongly positive patch tests, and the local dermatitis seen around infectious foci (infectious eczematoid dermatitis), may represent a limited form of this reaction.
Patients with a variety of infectious disorders may also present with an id reaction The most classic pattern is characterized by symmetrically distributed minute papules that have a predilection for the face, upper ears, and trunk. The most common causes are tinea capitis or an allergic contact dermatitis Therapy of tinea capi is with griseofulvin can lead to an id reaction soon after therapy that can be mistaken for an allergic reaction to griseofulvin. Proper management is to continue the griseofulvin and treat symptomatically with topical steroids or antihistamines if necessary. Id reactions can also be vesicular on the hands in response to an inflammatory tinea of the feet. Nummular eczematous lesions or pityriasis rosea–like lesions may occur in patients with head or pubic louse infestation. Id reactions clear when the focus of infection or infestation is treated, but topical or systemic antiinflammatory agents may be required until the triggering infection is eradicated.
The presence of a localized, chronic, and usually severe focus of dermatitis may affect distant skin in two ways. Patients with a chronic localized dermatitis may develop dermatitis at distant sites from scratching or irritating the skin. This is called ________
“conditioned irritability.”
The most common scenario is distant dermatitis in a patient with a chronic eczematous leg ulcer.
an eczematous disorder of children from age 3 years to puberty although rarely it persists into adulthood
Juvenile plantar dermatosis
It usually begins as a patchy, symmetric, smooth, red glazed patch on the base or medial surface of the great toes, sometimes with fissuring and desquamation. Unlike tinea, it spares the interdigital spaces. Lesions evolve into red scaling patches involving the weight-bearing and frictional areas of the feet, usually symmetrically. The skin ends up appearing like parchment paper and it fissures easily. The forefoot is usually much more involved than the heel. The eruption is disproportionately more common in atopic children. In some patients, a similar eruption occurs on the fingers. Histologically, there is psoriasiform acanthosis and a sparse, largely lymphocytic infiltrate in the upper dermis, most dense around sweat ducts at their point of entry into the epidermis. Spongiosis is often present, and the stratum corneum is thin but compact.
The disease is caused by the repeated maceration of the feet due to occlusive shoes often worn without socks, especially athletic shoes, sandals, or rubber shoes, or by the abrasive effects of pool surfaces or diving boards. Thin, nonabsorbent, synthetic socks may contribute to the problem, but socks that soak up excess water must be changed when soaked.
The diagnosis of plantar dermatosis is clinical, especially if there is a family or personal history of atopy and the toe webs are spared. Treatment involves avoidance of maceration. Foot powders, thick absorbent socks, absorbent insoles, and having alternate pairs of shoes to wear to allow the shoes to dry out are all beneficial. Topical corticosteroid medications are of limited value and often are no more effective than occlusive barrier protection. Petrolatum or urea preparations can sometimes be of benefit. Most cases clear within 4 years of diagnosis.
Allergic contact dermatitis may play a significant role in plantar dermatoses in childhood. In one study from Scotland, 50% of children with “inflammatory dermatitis” of the soles had relevant positive patch tests, and 4 of 14 children with typical juvenile plantar dermatitis also had a relevant contact allergen. Refractory plantar dermatitis in childhood should suggest allergic contact dermatitis.
dehydrated skin showing redness, dry scaling, and fine crackling that may resemble crackled porcelain or the fissures in the bed of a dried lake

Xerotic eczema, also known as winter itch, eczema craquelé, and asteatotic eczema. The primary lesion is an erythematous patch covered with an adherent scale. As the lesion enlarges, fine cracks in the epidermis occur (Fig. 5.14).
Fig. 5.14 Eczema craquelé.
Nummular lesions may occur. Xerotic “nummular” eczema is less weepy than classic nummular dermatitis. Favored sites are the anterior shins, extensor arms, and flank. Elderly persons are particularly predisposed, and xerosis is the most common cause of pruritus in older individuals. Xerotic eczema is seen most frequently during the winter, when there is low relative humidity. Bathing with hot water and harsh soaps contributes. The epidermal water barrier is impaired, and TEWL s increased. Epidermal barrier repair begins to decrease after age 55, correlated with an increase in epidermal pH (see later discussion). The loss of barrier repair ability is improved by acidifying the epidermis, showing the benefit of mild acids in treating xerosis. Heterozygous null mutation of the FLG gene is associated with xerosis.
Taking short tepid showers, limiting use of soap to soiled and apocrine-bearing areas, using acid pH synthetic detergents, and promptly applying an emollient after bathing are usually effective.
White petrolatum and emollients containing 10% urea or 5% lactic acid are effective. Topical corticosteroids in ointment vehicles are useful for inflamed areas.
Pruritic Dermatitis in Elderly Persons
Pruritic skin conditions are common in elderly patients, appearing around age 55 and increasing in severity with age. Males are more often affected, and Asians and Caucasians more frequently have pruritus as seniors than African Americans or Hispanics.
The dermatoses seen in this age-group are typically eczematous or papular. The eczematous plaques may resemble nummular dermatitis, a feature recognized by Marion Sulzberger when he coined the phrase “exudative discoid and lichenoid chronic dermatitis,” or “oid-oid disease.” The pathogenic basis of this component of dermatitis in elderly persons may be related to barrier failure due to loss of acidification of the epidermis. In addition, patients often have urticarial papules on the trunk and proximal extremities that resemble insect bites. These lesions are termed subacute prurigo and histologically demonstrate features of an arthropod assault, with superficial and deep perivascular lymphohistiocytic infiltrates, dermal edema, and at times interstitial eosinophils. Lesions of transient acantholytic dermatitis or eosinophilic folliculitis may also occur. This component of the eruption may be related to the tendency of elderly individuals to have an immune system that skews toward Th2 because of loss of Th1 function parallel to what occurs in the setting of AD. For this reason, some practitioners consider this “adult atopic dermatitis.” However, it is unknown whether these conditions have a genetic basis, or more likely, given the time of onset, are caused by acquired barrier and immune system abnormalities. In these patients, allergic contact dermatitis and photodermatitis may be present or develop. Patch testing may identify important allergens, avoidance of which leads to improvement.
Certain medication may also cause a similar eruption. Calcium channel blockers may be associated with pruritic dermatitis, but stopping them will clear only about one quarter of patients taking that class of medication. If there is widespread pruritic dermatitis, a biopsy should be performed to rule out cutaneous T-cell lymphoma. Treatment for these patients is similar to that of AD patients, with oral antipruritics, emollients, and topical corticosteroids (soak and smear) as first-line therapy. In refractory cases, phototherapy (UVB or PUVA), Goeckerman therapy (UVB plus crude coal tar) in a day treatment setting, and immunosuppressive agents can be effective. Inadvertent use of phototherapy in the patient with coexistent photosensitivity will lead to an exacerbation of pruritic dermatitis.
What are the primary lesions of Nummular Eczema (Discoid Eczema)?

Fig. 5.15 Nummular eczema. (Courtesy Steven Binnick, MD )
The primary lesions are discrete, coin-shaped, erythematous, edematous, vesicular, and crusted patches (Fig. 5.15).
NE usually begins on the lower legs, dorsa of the hands, or extensor surfaces of the arms. In younger adults, females predominate, but most patients older than 40 are male. Alcohol consumption has been associated with NE in adult males. A single lesion often precedes the eruption and may be present for some time before other lesions appear.
Most lesions are 2–4 cm in diameter. Lesions may form after trauma (conditioned hyperirritability). As new lesions appear, the old lesions expand as tiny papulovesicular satellite lesions appear at the periphery and fuse with the main plaque. In severe cases, the condition may spread into palm-sized or larger patches. Pruritus is usually severe and of the same paroxysmal, compulsive quality and nocturnal timing seen in AD and prurigo nodularis.
AD frequently has nummular morphology in adolescents, but in atopy the lesions tend to be more chronic and lichenified. Histologically, NE is characterized by acute or subacute spongiotic dermatitis. The skin lesions of nummular dermatitis are frequently colonized with S. aureus, in frequency similar to that seen in AD. Relevant positive patch tests are found in one quarter to one third of patients with NE. This may represent the primary cause of the dermatitis or a secondary allergy that developed from products used to treat the NE.
Management of nummular eczema
Initial treatment consists of simple soaking and greasing with an occlusive ointment, and once-daily or twice-daily application of a potent or superpotent topical corticosteroid cream or ointment then switched to a nonsteroidal topical medication such as a calcineurin inhibitor or crisaborole. Ointments are more effective, and occlusion may be necessary as nummular dermatitis can be very recalcitrant If secondary staphylococcal infection is present, an antibiotic with appropriate coverage can be used. Stopping alcohol consumption may improve response. A sedating antihistamine, doxepin, or gabapentin at bedtime can help with sleep and reduce nighttime scratching. In some cases refractory to topical agents, intralesional or systemic corticosteroid therapy may be required. In patients unresponsive to topical steroids, phototherapy with NB UVB, bath (soak), or oral PUVA can be effective. For refractory plaques, the addition of topical tar as 2% crude coal tar or 20% LCD may be beneficial
HORMONE-INDUCED DERMATOSES
Autoimmune progesterone dermatitis (APD) may appear as urticarial papules, deep gyrate lesions, papulovesicular lesions, an eczematous eruption, targetoid lesions, or a fixed drug reaction. Urticarial and erythema multiforme–like lesions are most characteristic. Lesions typically appear 5–7 days before menses, and improve or resolve a few days following menses Pruritus is common Onset is typically in the third and fourth decades of life. Familial cases have been reported. When urticaria is the predominant skin lesion, there is a generalized distribution, and it may be accompanied by laryngospasm and anaphylactoid reactions. Oral erosions may be present and cyclical, and a case of erythema multiforme associated with APD in an HIV patient has been reported. Many of the reported patients had received artificial progestational agents before the onset of the eruption. In some, it appeared during a normal pregnancy. The eruption may worsen
or clear during pregnancy. Rarely, it can occur in males given progesterone and adolescent females. Progesterone luteal-phase support during in vitro fertilization has exacerbated the disease.
In most cases, diagnosis has been confirmed by intradermal testing with progesterone. A positive test may be immediate (30 minutes) or delayed (24–96 hours). Flares may be induced by intramuscular, intravaginal, or oral progesterone. Histopathology shows a perivascular infiltrate in most with some interstitial involvement in a third and the infiltrate is mostly lymphocytes, but a minority of patients had eosinophils or neutrophils admixed. The most common treatment is an oral contraceptive to suppress ovulation, thereby reducing progesterone levels. Topical corticosteroids for mild eczematous cases and antihistamines in urticaria cases can be beneficial. For more refractory cases or in patients with erythema multiforme, fixed drug, or anaphylaxis as the cause, suppression of progesterone production with conjugated estrogen and gonadotropin-releasing antagonists such as leuprolide acetate, danazol, and tamoxifen has been successful. Desensitization protocols may allow for use of progesterone during in vitro fertilization and pregnancy. Menopause and oophorectomy (except in one reported patient) have been curative.
Autoimmune estrogen dermatitis also presents as a cyclic skin disorder that may appear eczematous, papular, bullous, or urticarial. Pruritus is typically present. Skin eruptions may be chronic but are exacerbated premenstrually or occur only immediately before the menses. Characteristically, the dermatosis clears during pregnancy and at menopause. Intracutaneous skin testing with estrone produces a papule lasting longer than 24 hours or an immediate urticarial wheal (in patients with urticaria). Injections of progesterone yield negative results, ruling out autoimmune progesterone dermatitis. Tamoxifen is effective in some cases.
IMMUNODEFICIENCY SYNDROMES presenting with skin manifestations
Primary immunodeficiency diseases (PIDs) are important to the dermatologist PIDs may present with skin manifestations, and the dermatologist may be instrumental in referring appropriate patients for immunodeficiency evaluation. These conditions have also given us tremendous insight into the genetic makeup and functioning of the immune system. The PIDs can be classified as those with predominantly antibody deficiency, impaired cell-mediated immunity (cellular immunodeficiencies, T cells, natural killer [NK] cells), combined B-cell and T-cell deficiencies, defects of phagocytic function, complement deficiencies, and wellcharacterized syndromes with immunodeficiency. More than 150
PIDs have been identified, as of the 2005 classification. Many of the original paradigms of PIDs have been refuted. PIDs are not rare, can be sporadic (not familial), can have adult onset, can be autosomal dominant, have incomplete penetrance, and may even spontaneously improve over time.
The dermatologist should suspect a PID in patients with chronic, severe, atypical or recalcitrant infections, and the type of immunodeficiency can at times be suggested by the clinical situation. Skin infections, especially chronic and recurrent bacterial skin infections, can be the initial manifestation of a PID with neutropenia, elevated IgE, or T-helper cell immunodeficiency. Fungal (especially Candida) and viral infections (warts, molluscum) suggest a PID of helper T cells or a specific monogenetic defect (STAT1 gain of function, IL-17, DOCK-8). Not all immunodeficiencies present with infec ions, but rather an inflammatory phenotype. Eczematous dermatitis and erythroderma, at times closely resembling severe atopic or seborrheic dermatitis, may affect the skin of PID patients due to maternal engraftment graft-versus-host disease (GVHD). They may be refractory to standard therapies. Granuloma formation, autoimmune disorders, and vasculitis are other cutaneous manifestations seen in some forms of primary immunodeficiency. The PIDs in which a specific infection or finding is the more common presentation are discussed in other chapters, including chronic mucocutaneous candidiasis (Chapter 15); Hermansky-Pudlak, Chédiak-Higashi, and Griscelli syndromes with pigmentary anomalies (Chapter 36); and cartilage-hair hypoplasia syndrome with disorders of hair (Chapter 27). The conditions described next are the most important PID conditions with which dermatologists should be familiar.
caused by mutations in the BTK gene (Bruton tyrosine kinase), which is essential for the development of B lymphocytes.
Bruton syndrome / X-linked agammaglobulinemia (XLA)
XLA typically presents between 4 and 12 months of life, when maternal immunoglobulins wane. The affected boys present with infections of the upper and lower respiratory tracts, gastrointestinal (GI) tract, skin, joints, and central nervous system (CNS). The infections are usually caused by Streptococcus pneumoniae, S. aureus, Haemophilus influenzae, Helicobacter, and Pseudomonas. Recurrent skin staphylococcal infection may be a prominent component of this condition. Atopic-like dermatitis and pyoderma gangrenosum have been described. Hepatitis B, enterovirus, and rotavirus infections are common in XLA patients, and one third develop a rheumatoid-like arthritis. Enterovirus infection may result in a dermatomyositis meningoencephalitis syndrome. Pyoderma gangrenosum has been reported in a patient, although this would absolutely be a diagnosis of exclusion because these patients can have such unusual skin infections. Kawasaki disease and polyarteritis nodosa have been reported in affected patients. An absence of palpable lymph nodes is characteristic.
Immunoglobulin A (IgA), IgM, IgD, and IgE are virtually absent from the serum, although IgG may be present in small amounts. The spleen and lymph nodes lack germinal centers, and plasma cells are absent from the lymph nodes, spleen, bone marrow, and connective tissues. In XLA, B cells usually only make up 0.1% of circulating peripheral blood lymphocytes (normal 5%–20%). More than 500 different mutations have been identified in the BTK gene in XLA patients. Some of these mutations only partially compromise the gene, so some patients may have milder phenotype and up to 7% circulating B cells, making differentiation from common variable immunodeficiency difficult. In addition to mutations in the BTK gene, mutations in other genes required for immunoglobulin production, such as IGHM, CD79A, CD79B, IGLLa, BLNK, and LRRC8A, can be responsible for panhypogammaglobulinemia.
Treatment with gamma globulin has enabled many patients to live into adulthood. The maintenance dose required can vary considerably from patient to patient. High-dose IVIG may also lead to improvement of pyoderma gangrenosum–like lower extremity ulcerations. Chronic sinusitis and pulmonary infection remain problematic because of the lack of IgA, and chronic sinopulmonary infections require repeated pulmonary function monitoring.
the most common immunodeficiency state
Isolated IgA Deficiency (OMIM 137100) - An absence or marked reduction of serum IgA (<7 mg/dL)
Patients with RAG-1 mutations can also present selective IgA deficiency. The incidence varies greatly based on ethnic background: about 1 : 150 in the Arab Peninsula and Spain, 1 : 225–1 : 300 in the United States, and 1 : 14,000–18,000 in Japan. Certain medications appear to induce selective IgA deficiency, including phenytoin, sulfasalazine, cyclosporine, nonsteroidal antiinflammatory drugs (NSAIDs), and hydroxychloroquine. The genetic cause in most cases is unknown.
From 10%–15% of all symptomatic immunodeficiency patients have IgA deficiency. Most IgA-deficient patients, however, are completely well. Of those with symptoms, half have repeated infections of the GI and respiratory tracts, and one quarter have autoimmune disease. Allergies such as anaphylactic reactions to transfusion or IVIG, asthma, and AD are common in the symptomatic group. There is an increased association of celiac disease, dermatitis herpetiformis, and IBD. Vitiligo, alopecia areata, and other autoimmune diseases (e.g., systemic lupus erythematosus [SLE], dermatomyositis, scleroderma, thyroiditis, rheumatoid arthritis, polyarteritis-like vasculitis, Sjögren syndrome) and ulcerative gingivitis have all been reported to occur in these patients. Malignancy is increased in adults with IgA deficiency.
heterogeneous disorder and is the most common immunodeficiency syndrome after IgA deficiency.

Common variable immunodeficiency (CVID)
Fig. 5.16 Common variable immunodefic ency with granulomas in vitiligo.
Patients have low levels of IgG and IgA, and 50% also have low levels of IgM. Lymphocyte counts may be normal or low. Multiple genetic defects have been found in CVID, including mutations in ICOS (CVID type 1), TNFRSF13B (type 2), CD19 (type 3), TNFRSF13C (type 4), MS4A1 (type 5), CD81 (type 6), CR2 (type 7), LRBA (type 8), PRKCD (type 9), and NFKB2 (CVID 10). These patients do not form antibodies to bacterial antigens, and have recurrent sinopulmonary infections. They have a predisposition to autoimmune disorders, such as vitiligo and alopecia areata, GI abnormalities, lymphoreticular malignancy (10-fold increase of lymphoma), and gastric carcinoma. Noninfectious granulomas have been reported in as many as 22% of CVID (Fig. 5.16) In some patients with CVID and multiple other immunodeficiencies, vaccine strain rubella has been found within the granulomas. Therefore it is hypothesized that the granulomas form due to difficulty processing the live rubella vaccination that is often given before diagnosis of CVID. Seven percent of CVID patients with granulomas have cutaneous granulomas, and virtually all patients with cutaneous granulomas also have visceral granulomas. These patients are more often female and have higher risk for lymphoma than other CVID patients. The granulomas can show multiple histologic patterns: granuloma annulare-like, sarcoidal, and even caseating. They show a CD4/CD8 ratio of less than 1, distinguishing these granulomas from sarcoidosis. CVID patients who develop granulomas have more severe depletion of isotype-switched memory B cells and naïve T cells, an immunologic profile also seen in ataxia telangiectasia patients with cutaneous granulomas Replacement of the reduced immunoglobulins with IVIG may help reduce infections. Topical, systemic, and intralesional corticosteroids may be used for the granulomas, depending on heir extent. Infliximab and etanercep have been effective in steroid-refractory cases.
This group of diseases includes defects that are combined T-cell and B-cell abnormalities, such as CD40 deficiency (CD40) and CD40 ligand deficiency (CD40LG), and disorders of primary B cells, such as cytidine deaminase (AICDA) and uracil-DNA glycosylase (UNG) deficiencies.
Class-Switch Recombination Defects (Formerly Immunodeficiency With Hyper-IgM)
Class-switch recombination defects are rare, and the different genetic diseases included in this group appear to have different clinical manifestations. These patients experience recurrent sinopulmonary infections, diarrhea, and oral and anogenital ulcers. Neutropenia may be associated with the ulcers. Recalcitrant human papillomavirus (HPV) infections (typically flat warts) may occur.
Hypomorphic mutations in NEMO or IKBKG are associated with hypogammaglobulinemia and elevated IgM and may be associated with anhidrotic ectodermal dysplasia with immunodeficiency. NEMO mutations cause X-linked recessive disorders with lymphocytosis and elevated CD3 and CD4 cells and low levels of NK cells. The mother may have mild stigmata of incontinentia pigmenti. These male infants present within the first few months of life with hypohidrosis, delayed tooth eruption, and immunodeficiency. Hair may be absent. Frequent infections of the skin and respiratory tract are common. The eruption has been characterized as an “atopic dermatitis–like eruption,” although some patients may have prominent intertriginous lesions resembling seborrheic dermatitis. Treatment is bone marrow transplantation.
occurs in adults in whom profound hypogammaglobulinemia and benign thymoma appear almost simultaneously
Thymoma with immunodeficiency, also known as Good syndrome
It is now classified predominantly as an antibody deficiency disorder. There is a striking deficiency of B and pre-B cells. One patient who developed vulvovaginal gingival lichen planus and severe oral herpes simplex has also been reported. Myelodysplasia and pure red blood cell aplasia may occur. Patients are at risk for fatal opportunistic pulmonary infections with fungi and Pneumocystis. Thymectomy does not prevent the development of the infectious or lymphoreticular complications. Supportive therapy with IVIG, granulocyte-macrophage colonystimulating factor (GM-CSF), and transfusions may be required.
an autosomal dominant disorder that in 50% of cases is caused by hemizygous deletion of 22q11-pter and rarely by deletions in 10p.
Digeorge Syndrome
Many cases are sporadic. Most DiGeorge syndrome patients have the congenital anomalies and only minor thymic anomalies. They present with hypocalcemia or congenital heart disease. The syndrome includes congenital absence of the parathyroids and an abnormal aorta. Aortic and cardiac defects are the most common cause of death. DiGeorge syndrome is characterized by a distinctive facies: notched, low-set ears, micrognathia, shortened philtrum, and hypertelorism. Patients with these DiGeorge congenital malformations and complete lack of thymus are deemed to have “complete DiGeorge syndrome.” Cell-mediated immunity is absent or depressed, and few T cells with the phenotype of recent thymus emigrants are found in the peripheral blood or tissues. Opportunistic infections are common despite normal immunoglobulin levels. Maternally derived GVHD may occur in these patients. A small subset of patients with complete DiGeorge syndrome develop an eczematous dermatitis, lymphadenopathy, and an oligoclonal T-cell proliferation. The condition may present as an atopic-like dermatitis, severe and extensive seborrheic dermatitis, or an erythroderma. This is called “atypical complete DiGeorge syndrome.” Biopsies show features of a spongiotic dermatitis with eosinophils, necrotic keratinocytes with satellite necrosis, and characteristically perieccrine and intraeccrine inflammation. This resembles the histology of grade 1 or 2 GVHD, lichen striatus, and some cases of mycosis fungoides. One African American patient with DiGeorge syndrome developed a granulomatous dermatitis. The treatment for complete DiGeorge syndrome is thymic transplantation.
A rare disorder called immune dysregulation, polyendocrinopathy, enteropathy, X-linked recessive (IPEX) syndrome presents neonatally with the classic triad of?
- autoimmune enteropathy,
- endocrinopa hy (diabetes, thyroiditis), and
- eczematous dermatitis affecting males.
Elevated IgE levels, eosinophilia, and food allergies, plus the eczematous dermatitis, all are manifestations of Th2 skewing of the immune system. Patients present with diffuse and severe erythematous exudative plaques resembling AD. Secondary infection is common, and staphylococcal septicemia can occur. The skin eruption may be follicularly based or may lead to prurigo nodularis. The scalp develops hyperkeratotic psoriasiform plaques. Cheilitis and onychodystrophy can occur. Alopecia areata, chronic urticaria, and bullous pemphigoid are cutaneous autoimmune manifestations of IPEX syndrome.
The IPEX syndrome is caused by mutations in FOXP3 (forkhead box P3 protein), the master control gene for regulatory T-cell (Treg) development. IPEX like disease may also be caused by loss-of-function mutations in CD25, STAT5b, and ITCH and gain-of-function mutations in STAT1 (signal transducer and activator of transcription 1). FOXP3 is necessary for the development of Tregs, which are required to maintain immune homeostasis and mediate peripheral tolerance to “self” and nonself antigens. The enteropathy may be driven by autoantibodies to villin, and these autoantibodies can be used diagnostically. Treatment is immunomodulator therapy or bone marrow transplantation.
heterogeneous group of genetic disorders characterized by severely impaired cellular and humoral immunity.
Severe combined immunodeficiency (SCID)
Severe T-cell deficiency and low lymphocyte count are found in virtually all SCID patients. Candidiasis (moniliasis) of the oropharynx and skin, intractable diarrhea, and pneumonia are the triad of findings that usually lead to the diagnosis of SCID. In addition, severe recurrent infections may occur, caused by Pseudomonas, Staphylococcus, Enterobacteriaceae, or Candida. Overwhelming viral infections are the usual cause of death. Engraftment of maternally transmitted or transfusion-derived lymphocytes can lead to GVHD. The initial seborrheic dermatitislike eruption may represent maternal engraftment GVHD. This cutaneous eruption may be asymptomatic but tends to generalize. Infants with a widespread dermati is but who lack any palpable lymph nodes should raise suspicion for SCID. More severe eczematous dermatitis and erythroderma may develop with alopecia. Cutaneous granulomas have been reported. In addition a higher rate of dermatofibroma sarcoma protuberans (DFSP) had been reported in SCID due to adenosine deaminase.
Deficiency or total absence of circulating T lymphocytes characterizes SCID. Immunoglobulin levels are consistently very low, but B-cell numbers may be reduced, normal, or increased. The thymus is very small; its malformed architecture at autopsy is pathognomonic.
The inheritance is either autosomal recess ve or X-linked. Forty percent of SCID cases are X-linked and caused by deficiency of a common γ-chain that is an essential component of the IL-2 receptor. Twenty percent are caused by adenosine deaminase (ADA) deficiency and 6% from Jak3 mutations.
Prenatal diagnosis and carrier detection are possible for many forms of SCID. The definitive treatment is hematopoietic stem cell transplantation (HSCT, bone marrow transplantation). This should ideally be carried out before age 3 1 2 months for optimal outcome. The success rate approaches 90%. In utero HSCT has been successful in X-linked SCID. SCID patients rarely live longer than 2 years without transplantation. On average, 8 years after successful HSCT, SCID patients may develop severe HPV infection with common warts, flat warts, or even epidermodysplasia verruciformis. The development of HPV infections in SCID patients after HSCT is only seen in patients with either Jak3 or γ-chain (gamma c) deficiency, but more than 50% of these patients may develop this complication.
Miscellaneous Genetic Disorders of Cellular Immunity
The TAP1 and TAP2 gene deficiencies are extremely rare autosomal recessive disorders that result in severe reduction of MHC class I expression on the surface of cells. CD8 cells are decreased, but CD4 cells are normal, as are B-cell numbers and serum immunoglobulins. Three forms of disease occur. The patient with the first phenotype develops severe bacterial, fungal, and parasitic infection and dies by age 3. The patient with the second phenotype is completely asymptomatic. The third group is the most common. Group 3 patients present in childhood with recurrent and chronic bacterial respiratory infections. These lead to bronchiectasis and eventually fatal respiratory failure in adulthood. The skin abnormalities appear in late childhood or more frequently in young adulthood (after age 15). Necrotizing granulomatous lesions appear as plaques or ulcerations on the lower legs and on the midface around the nose. The perinasal lesions are quite destructive and resemble “lethal midline granuloma” or Wegener granulomatosis. Nasal polyps with necrotizing granulomatous
histology also occur. One patient also developed leukocytoclastic vasculitis.
The ZAP-70 (ζ-chain [TCR]–associated protein kinase of 70 kD) deficiency is an autosomal recessive disorder of considerable heterogeneity. This enzyme is required for T-cell receptor (TCR) intracellular signaling. Patients present before age 2 years with recurrent bacterial, viral, and opportunistic infections, diarrhea, and failure to thrive. They have a lymphocytosis with normal CD4, NK, and B cells and decreased CD8 cells. Some patients develop an exfoliative erythroderma, eosinophilia, and elevated IgE levels.
Omenn syndrome (OMIM 603554; histiocytic medullary reticulosis) is a rare disorder that presents at birth or in the neo natal period. Classic Omenn was caused by defects in molecules involved in the variable diversity and joining V(D)J process. It is also caused by hypomorphic mutations in some of the genes that cause SCID. Both antibody production and cell-mediated immune function are impaired. Genetic mutations causing Omenn syndrome occur in RAG1 and RAG2 (90% of cases classic Omenn), DCLRE1C (encoding ARTEMIS), DNA-ligIV, IL7Rα, IL2Rγ, CHD7, ADA, and RNRP. These mutations all result in defective T-cell development and oligoclonal, abnormally activated T cells. Clinical features include severe exfoliative erythroderma, eosinophilia, alopecia, Pneumocystis jiroveci and viral pneumonias, colitis, hepatosplenomegaly, lymphadenopathy, hypogammaglobulinemia, and elevated IgE.
Wiskott-Aldrich syndrome, an X-linked recessive syndrome, consists of a triad of ______

Fig. 5.17 Eczematous eruption with purpura in Wiskott-Aldrich syndrome.
- chronic eczematous dermatitis resembling AD (Fig. 5.17);
- increased susceptibility to bacterial infections, such as pyoderma or otitis media; and
- thrombocytopenic purpura with small platelets.
- Levels of IgM are variable, IgA is normal to high, and IgE is elevated, as is IgG. On a complete blood cell count the mean platelet volume (MPV) is decreased. T cells (especially naïve T cells) are low in infancy and progressively decline in number and activity over time. Untreated survival is about 15 years, with death from infection, bleeding, or lymphoma (25% of patients).
The genetic cause of Wiskott-Aldrich syndrome is a mutation in what gene?
WASP gene
This gene codes for a protein called WASP, which is universally expressed in hematopoietic cells and is critical in the reorganization of the actin cytoskeleton in hematopoietic cells in response to external stimuli. The hematopoietic cells of affected patients cannot polarize or migrate in response to physiologic stimuli, accounting for the protean clinical features of the syndrome. Wiskott-Aldrich syndrome occurs when mutations in WASP lead to absence or truncation of the WASP protein (WASP − mutations). Mutations that result in normal length but some loss of function in the WASP protein (WASP + mutations) result in two different syndromes: X-linked thrombocytopenia (XLT) and intermittent X-linked thrombocytopenia. Gain-of-function mutations in WASP cause X-linked neutropenia. Patients with XLT may also have an atopic-like dermatitis, but this is usually milder than the severe and difficult to control eczema affecting patients with the full Wiskott-Aldrich syndrome. WASP/XLT patients may also develop autoimmune disease, especially autoimmune hemolytic anemia, vasculitis, Henoch-Schönlein–like purpura, and IBD. High IgM is associated with the development of autoimmune disease.
Treatment of wiskott-aldrich syndrome
Treatment is with platelet transfusions, antibiotics, and IVIG, if required. Low dose IL2 is in trials and targeted gene therapy has been attempted. Often, splenectomy is performed to help control bleeding, but this leads to increased risk of sepsis and is not routinely recommended. Immunosuppressive therapy or rituximab may be used to control autoimmune complications. Bone marrow transplantation from a human leukocyte antigen (HLA)–identical sibling as early as possible in the disease course provides complete reversal of the platelet and immune dysfunction, as well as improvement or clearing of the eczematous dermatitis. Survival at 7 years with a matched sibling donor transplant approaches 90%.
an autosomal recessive condition caused by mutations in a single gene on chromosome 11 (ATM), which encodes a protein called ATM
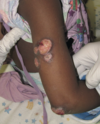
Ataxia Telangiectasia
Fig. 5.18 Granulomatous lesions in ataxia teleangiectasia.
When ATM is absent, the cell cycle does not stop to repair DNA damage, particularly double-stranded breaks, or for B(D)J recombination of immunoglobulin and TCR genes. This results in immunodeficiency and an increased risk for malignancy. The initial prominent skin feature is progressive ocular and cutaneous telangiectasias starting at age 3–6 years. These begin on the bulbar conjunctiva but later develop on the eyelids, ears, and flexors of the arms and legs. The ATM protein seems to be important in maintaining mitochondrial homeostasis, and this defect may be responsible for the premature aging (with loss of subcutaneous fat and graying of hair) and neurodegeneration.
Cutaneous noninfectious granulomas may occur and can be ulcerative and painful (Fig. 5.18). Rubella virus has been recovered by PCR from active ulcerations. Live vaccinations such the measles mumps and rubella vaccination should not be given to immunodeficient children, but because AT presents typically after age 1, children are often already vaccinated. Therefore the likely mechanism of granuloma formation is the ineffective processing of the rubella (or other infectious antigens) in live vaccinations. The granulomas tend to worsen with biopsy and manipulation. Other cutaneous features include large, irregular segmental café au lait spots, vitiligo, seborrheic dermatitis, AD, recurrent impetigo, and acanthosis nigricans. Late tightening of the skin can occur and resembles acral sclerosis. Sinopulmonary infections are common, especially otitis media, sinusitis, bronchitis, and pneumonia. Varicella (at times severe), herpes simplex, molluscum contagiosum, and herpes zoster can occur Refractory warts occur in more than 5% of patients. Aside from candidal esophagitis, unusual opportunistic infections are rare.
lls occurring in the majority of patients. Th-cell counts can be less than 200. IgA, IgG4, IgG2, and IgE deficiencies can all be present. Paradoxically, IgM, IgA, and IgG can be elevated in some patients, including the presence of monoclonal gammopathy in more than 10%. The immunologic abnormalities are not progressive. Lymphoma risk is increased more than 200-fold (especially B-cell lymphoma), and leukemia (especially T-cell chronic lymphocytic leukemia) is increased 70-fold. Treatment includes high vigilance for infection and malignancy. In patients with low CD4 counts, prophylaxis to prevent Pneumocystis pneumonia can be considered. When IgG deficiency is present and infections are frequent, IVIG may be beneficial. IVIG and intralesional corticosteroids may be used for the cutaneous granulomas. Carriers of ataxia telangiectasia have an increased risk for breast cancer. Because of the accumulation of chromosomal breaks after radiation exposure, both the ataxia telangiectasia patients and the carriers should minimize radiation exposure.
autosomal dominant disorder with hypogammaglobulinemia, reduced B-cell numbers, and neutropenia

The warts, hypogammaglobulinemia, infections, and myelokathexis (WHIM) syndrome
Fig. 5.19 Warts in WHIM syndrome
The most common genetic cause is a gain of function mutation of the CXCR4 gene. Additional mutations that are not in the CXCR4 gene can also cause WHIM, but all of them lead to functional hyperactivity of CXCR4. CXCR4 causes retention of neutrophils in the bone marrow and is the basis of the neutropenia and myelokathexis (increased apoptotic neutrophils in bone marrow). There is profound loss of circulating CD27+ memory B cells, resulting in hypogammaglobulinemia, with the observation that WHIM patients have normal antibody response to certain antigens but fail to maintain this antibody production. However, normal immunoglobulin levels do not exclude the diagnosis of WHIM. Almost 80% of WHIM patients have warts at the time of their diagnosis (Fig. 5.19). These include common and genital wart types. A significant number of female WHIM patients have cervical and vulval dysplasia, which can progress to carcinoma. WHIM patients have disproportionately more HPV infections than SCID patients but have little problem resolving other viral infections However, they may develop Epstein-Barr virus (EBV)induced lymphomas. The vast majority of patients in early childhood have recurrent sinopulmonary infections, skin infections, osteomyelitis, and urinary tract infections. Recurrent pneumonias lead to bronchiectasis. There is a CXCR4 antagonist in trials but standard treatment is G-CSF, IVIG prophylactic antibiotics, and aggressive treatment of infections. There is one report recently of a cure of WHIM by chromothripsis in a hematopoietic stem cell. Chromothripsis is a catastrophic disruption of chromosomes in a cell in this case the new stem cell had a selective advantage due to the lack of the WHIM mutation. Untreated, the HPV infections can progress to fatal carcinomas, and therefore male patients must be regularly examined by dermatologists and female patients by gynecologists; a low threshold for biopsy of genital lesions is required.
associated with hyper-IgE syndrome (see later discussion). However, unlike other genetic causes of hyper-IgE, this deficiency is uniquely associated with a susceptibility to cutaneous viral infections, including HSV, molluscum contagiosum, and HPV (Fig. 5.20).

DOCK8 Deficiency
Fig. 5.20 Warts in DOCK8 immunodeficiency
Warts can be flat or verrucous and affect about two thirds of patients.
important transcription factor involved in h topoiesis maintenance of the stem cell compartment
GATA2
GATA2 deficiency leads to a constellation of syndromes characterized by myelodysplasia, opportunistic infections, and leukemia. Patients have profound monocytopenia, often neutropenia, and NK, B, and dendritic cell lymphocytopenia. T-cell counts are variable. More than 75% of patients have severe or disseminated HPV infection, usually verruca plana or verruca vulgaris, and the HPV infection is first manifestation in the majority of patients, usually in adolescence or early adulthood. Severe cervical HPV infection can also occur and may lead to cancer. Thirty percent of patients develop a corticosteroid-responsive panniculitis. GATA2 has a role in lymphatic development and when the constellation includes lymphedema, it has been named WILD syndrome (warts, immunodeficiency, lymphedema, and dysplasia). The genetic defect in WILD patients can be missed on typical sanger sequencing because the mutation can be large and extend into exons. The WILD syndrome is rare and presents at age 6 months with lower extremity lymphedema that is progressive and later may involve the upper extremities and groin. Venous thrombosis occurs in 25% and lymphedema in 11% of patients of all patients with GATA2 mutations. Allogeneic HSCT seems to be curative
rare disorder caused by mutations in one of the genes that encode the subunits of the superoxide-generating phagocyte NADPH oxidase system responsible for the respiratory burst involved in organism killing
Chronic granulomatous disease (CGD)
CGD is characterized by repeated and recurrent bacterial and fungal infections of the lungs, skin, lymph nodes, and bones. Gingivostomatitis (aphthous-like ulcerations) and a seborrheic dermatitis of the periauricular, perinasal, and perianal area are characteristic. The dermatitis is frequently infected with S. aureus, and regional adenopathy and abscesses may complicate the infections. The term suppurative dermatitis is used in the immunology literature to describe this seborrheic-like dermatitis with secondary infection, analogous to the “infective dermatitis” seen in human T-cell lymphotropic virus (HTLV)–1 infection. In addition to S. aureus,
Serratia species are often isolated from skin abscesses and osteomyelitis. Aspergi lus is the most common agent causing pneumonia in CGD patients. In tuberculosis-endemic areas, CGD patients frequently develop active tuberculosis or prolonged scarring, abscesses, or disseminated infection following bacille CalmetteGuérin (BCG) immunization.
There are four types of CGD, one X-linked and three autosomal recessive. The X-linked form is the most common (65%–75% of CGD patients) and is caused by a mutation in the CYBB gene, which leads to a total absence of NADPH oxidase activity. In autosomal recessive forms, mutations in the genes encoding for the remaining three oxidase components have been described: p22-phox (CYBA), p47-phox (NCF1), and p67-phox (NCF2). One patient with a mutation in p40-phox (NCF4) has been described. The X-linked variant has the most severe phenotype. Compared with the autosomal recessive CGD patients, the X-linked patients present at an earlier age (14 vs. 30 months) and are diagnosed at an earlier age (3–5 vs. 6–13 years). The lack of superoxide generation apparently causes disease, not because the bacteria are not being killed by the superoxide, but because the superoxide is required to activate proteases in phagocytic vacuoles that are needed to kill infectious organisms.
Granuloma formation is characteristic of CGD and can occur in the GI tract, liver, bladder, bone, and lymph nodes. Up to 40% of biopsies from these organs will demonstrate granulomas, at times with identifiable fungal or mycobacterial organisms. These patients are often receiving prophylactic antibiotics, however, so organisms are frequently not found. Subcorneal pustular eruptions can also be seen in CGD patients. In the intestinal tract, the GI symptoms and granuloma formation can present in a similar fashion to inflammatory bowel disease.
The diagnosis of CGD is made by demonstrating low reduction of yellow nitroblue tetrazolium (NBT) to blue formazan in the “NBT test.” Dihydrorhodamine 123 flow cytometry (DHR), chemiluminescence production, and the ferricytochrome c reduction assay are also confirmatory. Western blot analysis for NADPH oxidase expression and DNA sequencing can pinpoint the genetic mutation.
Female carriers of the X-linked form of CGD have a mixed population of normal and abnormal phagocytes and therefore show intermediate NBT reduction and two discrete populations with DHR testing. The majority of carriers have skin complaints including a malar rash, discoid lupus erythematosus (DLE)–like lesions, adult acne and skin abscesses. The lesions are clinically DLE-like, but histologically, the interface component is often absent, and the lesions resemble tumid lupus. Direct immunofluorescence examination is usually negative, as is common in tumid lupus erythematosus (LE). Less frequently, CGD patients themselves have been described as having similar LE-like lesions or “arcuate dermal erythema.” Despite these findings, the vast majority of patients with LE-like skin lesions, both carriers and CGD patients, are antinuclear antibody (ANA) negative. Raynaud phenomenon can occur. More than half will report a photosensitive dermatitis, 40% have oral ulcerations, and one third have joint complaints.
Treatment of infections should be early and aggressive. There should be a low threshold to biopsy skin lesions, as they may reveal important and potentially life-threatening infections. Patients usually receive chronic TMP-SMX prophylaxis, chronic oral anti-Aspergillus agent (such as itraconazole, posaconazole or voriconazole), and IFN-γ injections. Voriconazole can lead to severe photodamage and skin malignancy, so chronic therapy in CGD patients should be used with extreme caution. Thalidomide has been used for the colitis. Bone marrow or stem cell transplantation has been successful in restoring enzyme function, reducing infections, and improving the associated bowel disease. However, survival is not increased with bone marrow transplantation, so this is not routinely undertaken. Recently IL-1 blockade was found to be effective at decreasing the inflammation of CGD.
Leukocyte adhesion deficiency (LAD) type I is caused by what mutation
Leukocyte adhesion deficiency (LAD) type I is caused by a mutation in the common chain (CD18) of the β2-integrin family (ITGB2).
It is characterized by recurrent bacterial infections of the skin and mucosal surfaces, especially gingivitis and periodontitis. The remnant umbilical cord is often delayed in separation during infancy Skin ulcerations from infection may continue to expand. Cellulitis and necrotic abscesses, especially in the perirectal area, can occur. Minor injuries may lead to pyoderma gangrenosum–like ulcerations that heal slowly. Infections begin at birth, and omphalitis with delayed separation of the cord is characteristic. Neutrophilia is marked, usually 5–20 times normal, and the count may reach up to 100,000 during infections. Despite this, there is an absence of neutrophils at the sites of infection, demonstrating the defective migration of neutrophils out of the blood vessels in these patients. LAD type I patients are affected either severely (<1% of normal CD18 expression) or moderately (2.5%–10% of normal expression.) Patients with moderate disease have less severe infections and survive into adulthood, whereas patients with severe disease often die in infancy
LAD type II is caused by what mutation?
LAD type II is caused by a mutation in SLC35C1, which results in a general defect in fucose metabolism which results in decreased fucosylation of selectin ligands on leukocytes. This leads to impaired tethering and rolling on activated endothelial cells. Severe mental retardation, short stature, a distinctive facies, and the rare hh blood phenotype are the features. Initially, these patients have recurrent cellulitis with marked neutrophilia, but the infections are not life threatening. After age 3 years, infections become less of a problem and patients develop chronic periodontitis.
LAD type III is caused by what mutation?
LAD type III is caused by a mutation in the gene FERMT3 and is characterized by severe recurrent infections, bleeding tendency (from impaired platelet function), and marked neutrophilia.
LAD1 patients can have abnormal IL12 and 23 activity and thus ustekinumab was recently tried with excellent success Bone marrow transplantation is an option for patients with severe LAD type I and LAD type III.
The autosomal dominant form of hyperimunoglobulinemia E syndrome (HIES; also called hyper-IgE syndrome) is caused by what mutation?
STAT3
Autosomal dominant HIES was first called Job syndrome or Buckley syndrome. The classic triad is an AD-like eczematous dermatitis, recurrent skin and lung infections, and high serum IgE, although IgE is elevated in atopy generally. The skin disease is the first manifestation of STAT3 deficiency and begins at birth in 19% of cases, within the first week of life in more than 50%, and in the first month in 80%. This is very helpful clinically because AD does not usually start until the second or third month of age. The initial eruption is noted first on the face or scalp, but quickly generalizes to affect the face, scalp, and body. The rash favors the shoulder, arms, chest, and buttocks. The newborn rash begins as pink papules and pustules that coalesce into crusted plaques that may initially be diagnosed as “neonatal acne.” Histologically, these papules are intraepidermal eosinophilic pustules. The dermatitis evolves to bear a close resemblance to AD, often very severe, and occurs in 100% of autosomal dominant HIES patients. Staphylococcal infection of the dermatitis is common. There can be “cold” abscesses due to the lack of inflammation typically present to fight infection. IgE levels are above 2000 in 95% of patients with autosomal dominant HIES, but since only a small percentage of children with IgE levels above 2000 actually have HIES, other features must be used to confirm the diagnosis. Recurrent pyogenic pneumonia is the rule, starting in childhood. Because of the lack of neutrophilic inflammation in abscesses and pneumonia, symptoms may be lacking and lead to a delay in diagnosis. Although antibiotic treatment clears the pneumonia, healing is abnormal, with the formation of bronchiectasis and pneumatoceles, a characteristic feature of HIES. Mucocutaneous candidiasis is common, typically thrush, vaginal candidiasis, and candidal onychomycosis. Musculoskeletal abnormalities are common, including scoliosis, osteopenia, minimal trauma fractures (55%), and hyperextensibility, leading to premature degenerative joint disease. Retention of some or all of the primary teeth is a characteristic feature. Other oral manifestations include median rhomboid glossitis, high-arch palate, and abnormally prominent wrinkles on the oral mucosa. Arterial aneurysms are common, including Chiari 1 malformation (40%) and coronary vascular abnormalities (60%). The latter can cause myocardial infarction. Autosomal dominant HIES patients have a characteristic facies, developing during childhood and adolescence. Features include facial asymmetry, broad nose, deep-set eyes, and a prominent forehead. The facial skin is rough, with large pores. There is an increased risk of malignancy, predominantly B-cell non-Hodgkin lymphoma, but cutaneous squamous cell carcinoma has also been reported. Laboratory abnormalities are limited to eosinophilia and an elevated IgE. In adults, IgE levels may become normal. Functional Th17 studies can help in diagnosis. A scoring system developed at the National Institutes of Health (NIH) can accurately identify patients with HIES, selecting those in whom genetic testing could be considered.
The autosomal recessive form of hyperimunoglobulinemia E syndrome (HIES; also called hyper-IgE syndrome) is caused by what mutation?
DOCK8 and rarely in tyrosine kinase 2 (TYK2) or phosphoglucomutase 3 (PGM3)
GM3 mutations and is less common. Patients with DOCK8 mutations also have severe eczema and recurrent skin and lung infections, although the lung infections resolve without pneumatoceles (except in PGM3). TYK2 has only been described in a small number of patients who had high IgE and atopy. Food allergies are often present in autosomal recessive HIES caused by DOCK8 mutation, as is decreased IgM. The main cutaneous difference is patients with DOCK8 mutations are predisposed to exuberant cutaneous viral infections, especially warts, molluscum contagiosum, herpes s mplex, and varicella-zoster, as well as systemic viral infections, including EBV and cytomegalovirus (CMV) They also develop mucocutaneous candidiasis. Neurologic disease is much more common in autosomal recessive HIES, ranging from facial paralysis to hemiplegia. Autosomal recessive HIES patients have normal facies, no fractures, and normal shedding of primary dentition, but a dramatic increase in malignancy, especially leukemia.
Management of HIES?
Treatment for HIES is aimed at decreasing infection and maintaining skin barrier. Infections are suppressed with bleach baths and chronic antibiotic prophylaxis (usually with TMP-SMX); antifungal agents may be used for candidal infections of the skin and nails. Topical antiinflammatories are used to manage the eczema, and in severe cases, systemic therapy can be considered but should be used with caution. Bisphosphonates are used for osteopenia. The role of IVIG, antihistamines, dupilumab, and omalizumab (antibody against IgE) is unknown. In patients with autosomal recessive HIES, hematopoietic cell transplantation (HCT) is recommended because of the high risk of malignancy and CNS infarction. Autosomal dominant HIES patients with malignancy should be considered for HCT because it can reverse the HIES, reducing the infectious complications following HCT.
Four major functions that result from complement activation
cell lysis, opsonization/ phagocytosis, inflammation, and immune complex removal
In the “classical” complement pathway, complement is activated by an antigen-antibody reaction involving IgG or IgM. Some complement components are directly activated by binding to the surface of infectious organisms; this is called the “alternate” pathway. The central component common to both pathways is C3. In the classical pathway, antigen-antibody complexes sequentially bind and activate three complement proteins, C1, C4, and C2, leading to the formation of C3 convertase, an activator of C3. The alternate pathway starts with direct activation of C3. From activated C3, C5–C9 are sequentially activated. Cytolysis is induced mainly through the membrane attack complex (MAC), which is made up of the terminal components of complement. Opsonization is mainly mediated by a subunit of C3b, and inflammation by subunits of C3, C4, and C5.
Inherited deficiencies of complement are usually autosomal recessive traits. Deficiencies of all 11 components of the classical pathway, as well as inhibitors of this pathway, have been described. Genetic deficiency of the C1 inhibitor is the only autosomal dominant form of complement deficiency and results in hereditary angioedema (see Chapter 7). In general, deficiencies of the early components of the classical pathway result in autoimmune connective tissue diseases, whereas deficiencies of the late components of complement lead to recurrent neisserial sepsis or meningitis. Overlap exists, and patients with late-component deficiencies may exhibit connective tissue diseases, and patients with deficiencies of early components, such as C1q, may manifest infections. Deficiency of C3 results in recurrent infections with encapsulated bacteria such as Pneumococcus, H. influenzae, and Streptococcus pyogenes. C3 inactivator deficiency, as with C3 deficiency, results in recurrent pyogenic infections Properdin (component of alternate pathway) dysfunction is inherited as an X-linked trait and predisposes to fulminant meningococcemia. Deficiency of C9 is the most common complement deficiency in Japan but is uncommon in other countries Most patients appear healthy. MASP2 deficiency, resulting in absent hemolytic activity by the lectin pathway, is considered a complement deficiency and results in a syndrome resembling SLE and increased pyogenic infection. Factor I deficiency results in recurrent infections, including Neisseria meningitides. Partially deficient family members may also have increased infections.
C2 deficiency is the most common complement deficiency in the United States and Europe Most patients are healthy, but SLE-like syndromes develop in 10%. C1q-, C3-, and C4-deficient patients have SLE at rates of 90%, 31%, and 75%, respectively, and C1r deficiency was recently reported in early-onset SLE. Complement deficiency–associated SLE typically has early onset, photosensitivity less renal disease, and Ro/La autoantibodies in two thirds of patients. C2- and C4-deficient patients with LE typically have subacute annular morphology (Fig 5.21), Sjögren syndrome, arthralgias, and oral ulcerations. CH50 is a functional assay of the complement system and is typically low in active systemic lupus. Cell-bound complement activity products such as C4d and C3d, which are the byproducts of complement activation, are deposited on other cells such as erythrocytes and can be detected to monitor systemic lupus disease activity. Frequent infections, anaphylactoid purpura, dermatomyositis, vasculitis, and cold urticaria may be seen. A patient with C1q deficiency presented with macrophage activating syndrome. Renal disease, anti-dsDNA antibodies, and anticardiolipin antibodies are uncommon. Patients with C4 deficiency may have lupus and involvement of the palms and soles.
Many of the complement component deficiencies can be acquired as an autoimmune phenomenon or a paraneoplastic finding. Examples include acquired angioedema, as when C1 inhibitor is the target, or lipodystrophy and nephritis, when C3 convertase is the target.
When complement deficiency is suspected, a useful screening test is a CH50 (total hemolytic complement) determination, because deficiency of any of the complement components will usually result in CH50 levels that are dramatically reduced or zero.
Fig 5 21 Subacute lupus erythematosus as can be seen in complement deficiency disorders.

Graft-Versus-Host Disease
GVHD occurs most frequently in he setting of HSCT but may also occur following organ transplantation or in the rare situation of transfusion of active lymphoid cells into an immunodeficient child postpartum or even in utero. Blood transfusions with active lymphocytes (nonradiated whole blood) from family members or in populations with minimal genetic variability, given to an immunodeficient patient, can result in GVHD. HSCT from a monozygotic twin (syngeneic) or even from the patient’s own stem cells (autologous) can rarely induce a mild form of GVHD.
What are the 3 elements required for the development of GVHD?
- First, the transplanted cells must be immunologically competent.
- Second, the recipient must express tissue antigens that are not present in the donor and therefore are recognized as foreign.
- Third, the recipient must be unable to reject the transplanted cells.
Immunologic competence of the transplanted cells is important, because ablating them too much may lead to failure of engraftment, or, more often, incomplete eradication of the recipient’s malignancy (graft-vs.-tumor effect). Therefore some degree of immunologic competence of the transplanted cells is desired. For this reason, the prevalence of GVHD still remains about 50% after HSCT. Another important factor in determining the development and severity of the GVHD is the preconditioning regimen. Chemotherapy and radiation cause activation of dendritic cells (antigenpresenting cells [APCs]) in tissues with high cell turnover—the skin, gut, and liver. These APCs increase their expression of HLA and other minor cell surface antigens, priming them to interact with transplanted lymphoid cells. Host APCs are important in presenting these antigens to the active lymphoid donor cells. Cytokines, especially IL-2, TNF-α, and IFN-γ, are important in enhancing this host-donor immunologic interaction. Reducing this early inflammatory component in GVHD can delay the onset of the GVHD but may not reduce the prevalence. The indications for HSCT, age limits, and allowable degree of HLA incompatibility have resulted in greater use of HSCT, increasing the number of persons at risk for GVHD.
Initially, only reactions that occurred within the first 100 days after transplantation were considered acute GVHD, but it is now recognized that classic acute GVHD can occur up to 1 year or more after HSCT, especially with tapering of anti-GVHD immunosuppressives. Acute GVHD is based on the clinical presentation, not the time of onset after transplantation. In acute GVHD, the cutaneous eruption typically begins between the 14th and 42nd days after transplantation, with a peak at day 30 (Fig. 5.22). Acu e GVHD is characterized by an erythematous morbilliform eruption of the face and trunk, which may become confluent and result in exfoliative erythroderma. Ear involvement is common. It often begins with punctate lesions corresponding to hair follicles and eccrine ducts, resembling keratosis pilaris. In many cases, a fine scale can be appreciated on the skin lesions. GVH is often described as more monomorphous than similar-appearing morbilliform drug eruptions, and tends to involve the upper back, rather than the lower back and dependent areas. GVHD is staged clinically, with stage 1 as less than 25% BSA, stage 2 as 25%–50% BSA, stage 3 as greater than 50% BSA, and stage 4 as erythroderma with bullae. In children, the diaper area is often involved The eruption may appear papular and eczematous, involving web spaces, periumbilical skin, and ears, and bears some resemblance to scabies.
The differential diagnosis for the eruption of acute GVHD includes the eruption of lymphocyte recovery, engraftment syndrome, viral exanthem, and drug eruption. The cutaneous histology in the early phases of acute GVHD may not be able to completely distinguish these entities. Grade IV GVHD is characterized by full-thickness slough and may resemble toxic epidermal necrolysis, and it may be impossible to distinguish the two entities clinically or histologically. The mucous membranes and the conjunctivae can be involved as well, which can be difficult to distinguish from chemotherapy-induced and infectious mucositis. Often, about the same time, the patient develops the other characteristic features of acute GVHD: cholestatic hepatitis with elevated bilirubin and high-volume diarrhea Autologous GVHD usually involves only the skin and is self-limited, and it is thought to occur from preconditioning regimen–induced loss of “self-tolerance.”
The “eruption of lymphocyte recovery” is a mild, self-limited morbilliform eruption that may be associated with a brief fever, occurring when lymphocyte counts first return following chemotherapy. This can closely resemble GVHD, though without liver or gut involvement; patients may progress to overt aGVHD. Engraftment syndrome is a combination of symptoms that occur about the time of engraftment and neutrophil recovery. Patients
develop fever (without infectious source), diarrhea, pulmonary infiltrates with hypoxia, and capillary leak syndrome with edema and weight gain. It occurs as soon as 7 days after autologous HSCT and 11–16 days after allogeneic transplants. The associated skin eruption is clinically and histologically identical to acute GVHD. Ocular involvement with keratitis can occur. This syndrome occurs in 7%–59% of post-HSCT patients and is a significant cause of morbidity and mortality in autologous peripheral blood progenitor cell transplant patients. It is mediated by cytokine production and neutrophil infiltration of the organs damaged by the conditioning chemotherapy, especially the lungs. Administration of G-CSF and autologous transplantation are risk factors for its development. Treatment is high-dose systemic corticosteroids.
With improved support for GVHD patients after HSCT, more are surviving, and 60%–70% develop chronic GVHD (cGVHD). It is the second most common cause of death in HSCT patients. It is unclear whether cGVHD is mediated by the same pathologic mechanisms as acute GVHD. Certain conditioning regimens may increase risk of cGVHD, particularly total body irradiation and the risk of scleroderma-like cGVHD. Chronic disease has features more typical of an autoimmune disease. Diagnostic criteria have been adopted, with “diagnostic” and “distinctive” cutaneous manifestations. The most common diagnostic feature, occurring in 80% of patients who develop cGVHD, is a lichen planus–like eruption. It typically occurs 3–5 months after grafting, usually beginning on the hands and feet but becoming generalized It may present with a malar rash resembling LE. The chronic interface dermatitis can leave the skin with a poikilodermatous appearance. Similar lichen planus–like lesions may occur on the oral mucosa and can result in pain and poor nutrition. Lichen sclerosus–like lesions can also occur. Involvement of the vaginal or esophageal mucosa can result in severe scarring and strictures. About 20% of men with cGVHD have genital skin changes, and 13% have cGVHD of the penis. cGVHD of the skin and oral mucosa is associated with genital involvement. Lichen sclerosus–like lesions, phimosis, and inflammatory balanitis are most common; 80% of men with penile cGVHD report erectile dysfunction. Patients may underreport their genital cGVH, and a thorough physical and history is important.
Fig. 5.22 Acute graft-versus-host disease. (Courtesy Jennifer Huang, MD.)
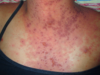
Fig. 5.23 Chronic graft-versus-host disease.

Sclerosis is the other “diagnostic” family of skin lesions. This can include lesions resembling superficial morphea, which can have overlying lichen sclerosus–like changes. The morphea-like lesions demonstrate an isomorphic response, favoring areas of pressure, especially the waistband and brassiere-band areas. Deeper sclerotic lesions resembling eosinophilic fasciitis (resulting in joint contractures, Fig. 5.23) and restriction of the oral commissure due to sclerosis can occur. These sclerotic plaques may ulcerate.
The extent of involvement of the deep tissues, such as muscle and fascia, cannot be easily defined by clinical examination and may be aided by magnetic resonance imaging. Rarely, the myositis of cGVHD may be accompanied by a skin eruption similar to dermatomyositis. Other features of GVHD may include depigmentation resembling vitiligo; scarring or nonscarring alopecia; nail dystrophy (e.g., longitudinal ridging, brittle thin nails, pterygium, nail loss); and xerostomia and other, Sjögren-like mucosal symptoms. Atypical forms of GVHD resembling Pityriasis Rubra Pilaris (PRP), psoriasis, eczematous eruptions, isolated follicular hyperkeratosis, and Pityriasis Rosea (PR)-like eruptions have been reported. GVHD associated angiomatosis, with vascular lesions within sclerotic-type cGVHD, is a newly described and challenging subtype of cGVHD.
Biopsy may not be necessary to diagnose GVHD, particularly aGVHD. Punch biopsy is generally adequate, unless evaluating scleroderma-like disease, where incisional biopsy is preferred. Histologically, aGVHD demonstrates vacuolar interface dermatitis. Individual keratinocyte necrosis with adjacent lymphocytes (satellite necrosis) is typically present, suggesting cell-mediated cytotoxicity. The height and extent of keratinocyte necrosis, bulla formation, and slough are used in grading schemes (grade 0 is normal, grade 1 is basal vacuolization, grade 2 includes necrotic epidermal cells, grade 3 has clefting, and grade 4 has bulla formation). Eosinophils may be more common in drug reactions, but can be seen in GVH and cannot be used to distinguish the two. GVHD inflammation histologically often involves follicles, corresponding to the characteristic clinical perifollicular inflammation. Elafin has been proposed as a possible marker for GVHD but cannot reliably distinguish GVHD from drug reactions, though high levels of elafin may correspond to a poorer prognosis. The histologic findings in early disease may be nonspecific, and many treatment protocols do not depend on histologic features to initiate therapy. A background of epidermal disorder and atypia may be present in later lesions of acute GVHD, but similar epidermal changes may be seen after chemotherapy and may be unreliable in distinguishing GVHD from drug reactions. Chronic GVHD demonstrates lichenoid dermatitis or dermal sclerosis with hyalinization of collagen bundles and narrowing of the space between bundles.
Acute GVHD is managed on the skin with topical corticosteroids, TCIs, and rarely UV phototherapy. Most patients have associated systemic symptoms, in which case systemic corticosteroids, tacrolimus, and occasionally other suppressive agents are instituted. When aGVH is not responsive to steroids, multidisciplinary discussions and individual regimens based on comorbidities, infectious history, and status of underlying disease are often necessary. Mesenchymal stem cells, antithymocyte globulin, and extracorporeal photopheresis (ECP) are novel options being evaluated. TNF inhibitors, rituximab, and targeted small molecule inhibitors are also being studied. Ruxolitinib and other JAK inhibitors have shown promise in early studies and are being actively evaluated for both acute and chronic GVHD. For cGVHD, ECP can be considered as a nonsuppressive, disease-stabilizing, steroid-sparing agent for patients unresponsive to these first-line therapies. Phototherapy, particularly UVA-based regimens (PUVA with or without retinoids, UVA1), may improve sclerotic cGVHD. Rituximab has shown some benefit in steroid-refractory cGVHD. Sirolimus and everolimus appear to have activity against fibrosis and may be useful in fibrotic cGVHD. Imatinib may also be useful in cGVHD with fibrosis. Hydroxychloroquine has also been reported. In cGVHD, topical steroids may help lichen planus–like disease but are unlikely to affect scleroderma-like disease. Physical therapy is essential in patients with sclerosis, especially if it involves joints.
Graft-Versus-Host Disease in Solid-Organ Transplantation
Transplantation of a solid organ into a partially immunosuppressed host may result in GVHD, because the organ may contain immune cells. The prevalence of GVHD after solid-organ transplantation is extremely low, about 1% at one center over 15 years with more than 2000 transplants. The risk for developing GVHD after solid-organ transplantation is related to the type of organ transplanted and depends on the amount of lymphoid tissue that the organ contains. The risk profile is small intestine > liver/pancreas > kidney > heart. In liver and small intestine transplants the risk is 1%–2%, but when it occurs the mortality rate is 85%. Close matching increases the risk of GVHD in organ transplantation, because the immunocompetent recipient cells are less likely to recognize the donor lymphocytes as nonself and destroy them. Also, African American race and CMV infection increase the risk The onset is usually 1–8 weeks following transplantation but can be delayed for years. Fever, rash, and pancytopenia are the cardinal features, with pancytopenia a frequent cause of mortality. The skin is the first site of involvement, and only cutaneous disease occurs in 15% of cases. Both acute and chronic GVHD skin findings can occur. Skin biopsies tend to show more inflammation than in HSCT-associated GVHD. In GVHD accompanying liver transplantation, the liver is unaffected because it is syngeneic with the donor lymphocytes. The diagnosis of GVHD in patients receiving organ transplantation can be aided by documenting macrochimerism in the peripheral blood and skin after the first month of transplantation.
