Khayal - Ant Pituitary/Adrenal Disorders Flashcards
Why does the hypothalamus represent the coordinating center of the endocrine system?
- Delivers precise signals to pituitary gland, which releases hormones that affect most endocrine systems in the body
- Hypothalamus consolidates signals from upper cortical centers, autonomic function, environmental cues and peripheral feedback
How is the vascular supply of the anterior pituitary unique?
- Anterior pituitary lacks major arterial blood supply, making it susceptible to infarction (Sheehan’s Syn)
- Dense capillary network of pituitary portal blood/plexus containing hypothalamic hormones and the hormones/paracrine/autocrine factors released form the pituitary cells
- Venous plexus empties into petrosal sinuses, then into the peripheral circulation via the jugular veins
- In short, this is an exception to the standard vascular supply: venous - capillaries - venous (hypothalamic-hypophyseal portal veinous system)

What is the embryological origin of the anterior and posterior pituitary?
- Anterior pituitary is derived from the foregut
- Posterior is derived from neural tissue
Describe the vascular and neural connections bt the hypothalamus and pituitary.
- PV nucleus: neurons w/terminals in the median eminence, where hormones are released directly into primary capillary plexus, traverse long portal veins in the pituitary stalk, and enter secondary plexus in the anterior pituitary
- Hypothalamic-hypophyseal portal veinous system: primary plexus, long portal venous system, and secondary plexus
- SA nucleus: axons travel down supraopticohypophyseal tract and release ADH and oxytocin directly in the posterior pituitary
- NOTE: this distinction b/t nuclei not important; just understand the uniqueness of the vascular system

What are the hypothalamic stimulatory and inhibitory hormones? Pituitary correlates?
- CRH: POMC products -> ACTH, MSH, and endorphins (20% of ant pit cells; basophilic)
- GHRH: GH (50% of ant pit cells; acidophilic)
- GnRH: LH and FSH (15% ant pit cells; basophilic)
- TRH: TSH (5% ant pit cells; basophilic)
- PRH (serotonin, ACTH, opiates, estrogen): prolactin (10-30% ant pit cells; acidophilic)
- Somatostatin: INH release of growth hormones
- Dopamine: INH of prolactin release
What is hypopituitarism? What are 2 common causes?
- DEC secretion of pituitary hormones that can result from diseases of pituitary or hypothalamus
• Clinical manifestations depend on the cause and the type of hormonal deficiency
- Pituitary surgery is the most common cause of hypopituitarism
- Sheehan syndrome: pituitary hypertrophies during pregnancy, blood supply does not -> hypotensive at birth (due to hemorrhage), infarcting the pituitary
What are the major causes of hypopituitarism?
- PITUITARY DISEASE: mass lesions (pituitary adenomas, benign tumors, cysts, etc.), pituitary surgery, radiation, infiltrative lesions (lymphocytic hypophysitis, hemochromatosis), infarction (Sheehan syndrome), apoplexy, genetic muts (Pit-1)
- HYPOTHALAMIC DISEASE: mass lesions (benign - craniopharyngioma, malignant - metastatic from lung, breast), radiation (for CNS, nasopharyngeal malignancies), infiltrative lesions (sarcoidosis, langerhans cell histiocytosis), trauma (fracture of skull base), infectious (TB meningitis)
What are the clinical features of hypopituitarism?
- Varies from asymptomatic to acute collapse, depending on etiology, rapidity of onset, and predominant hormones involved
1. ACTH def: adrenal insufficiency
2. TSH def: hypothyroidism
3. Gonadotropin def: hypogonadism
4. GH def: failure to thrive, short stature in kids; most adults asymptomatic, but some may have fatigue, weakness, and DEC QOL
5. Prolactin def: failure of lactation
6. ADH def: diabetes insipidus (polyuria and polydipsia)
What is the treatment for hypopituitarism?
Includes a sum of the tx for each of the individual pituitary hormone deficiencies
What is growth hormone? When/how is it secreted?
- Main hormone regulating growth -> rapid growth in puberty, for example
- Secreted in a PULSATILE FASHION
- Stimulation from the hypothalamus via GHRH, and INH by Somatostatin (also secreted by hypothalamus)
- Starvation and hypoglycemia are 2 potent stimuli for secretion

What 2 conditions can GH excess cause?
- Gigantism: growth plates are not fused, resulting in extraordinarily tall stature and other signs of GH excess
- Acromegaly: growth plates are fused, so other abnormalities of GH will be present
What is acromegaly?
- Persistent hypersecretion of GH (most common cause a sematotroph adenoma)
-
Insidious onset & slow progression: avg interval from onset of symptoms to dx 12 years
1. At dx, 75% have macroadenomas (>10mm), and some extend to para- or suprasellar areas - Clinical features due to high serum concentrations of GH and and IGF-1 -> somatic/metabolic effects
1. Metabolic effects include: nitrogen retention, insulin antagonism, lipolysis
What are the causes of acromegaly?
-
Primary GH excess: GH-cell adenoma, mixed GH-PRL-cell adenoma, Mammosomatotroph-cell adenoma, Plurihormonal adenoma, GH-cell carcinoma
1. Familial syndromes: MEN-1, Familial acromegaly, McCune-Albright, Carney’s - Ectopic/iatrogenic GH excess: pancreatic islet-cell tumor, lymphoma, iatrogenic
- GHRH excess: central ectopic (<1%: hypothalamic hamartoma, choristoma, ganglioneuroma), peripheral ectopic (1%: bronchial carcinoid, small cell lung cancer, adrenal adenoma, pheochromocytoma)

What are the clinical manifestations of acromegaly?
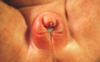
- Direct tumor affects: headache/vision loss (bitemporal hemianopsia: see attached image)
- Pituitary function: macroadenoma can cause pituitary hormone deficiency by gland compression (most commonly, gonadotropin deficiency)
- Mortality rate of these pts INC, esp. if strict biochemical control not achieved
1. INC CARDIOVASCULAR DISEASE mortality

What are the presenting clinical features of acromegaly?
- More than 50% present with:
1. Acral enlargement
2. Maxillofacial changes

What are the risks of long-term exposure to GH excess?
- Arthropathy: rapid symptomatic improvement, but irreversible bone and cartilage lesions (unrelated to age or GH secretion: usually w/long duration)
- Neuropathy: peripheral anesthesias, paresthesias, and sensorimotor polyneuropathy, onion bulbs do NOT regress
- CV: CARDIOMYOPATHY, arrhythmias (LV mass INC, diastolic func DEC), fibrous CT hyperplasia
- HTN: exacerbates cardiomyopathy, may progress even if GH secretion reduced
- Pulm: upper airway obstruction via soft tissue overgrowth (improves w/reduced GH secretion)
- Malignancy: INC risk, INC colon polyps, effect of therapy on risk unknown
- Carb intolerance: DM, improves w/DEC secretion
- NOTE: MOST OF THESE IMPROVE WITH TX
How does acromegaly affect your risk of malignancy?
INC it

What are the major diagnostic features of acromegaly?
- SYMPTOMS: headache, heat intolerance, ring and shoe sizes, facial bony changes
- SIGNS: prominent forehead, broad nose, prominent lower jaw, visual field loss
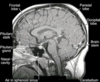
- MRI: commonly, macroadenoma
- BIOCHEM: elevated IGF-1, GH nadir >2 after oral glucose dose
- PATHO: GH-staining pituitary adenoma

How do you diagnose pt with acromegaly?
- Measurement of IGF-1 (NOT GH)
-
OGTT: in normal subjects GH levels suppress to <1 ng/ml 2 hours after ingestion of 75 g of glucose
1. In acromegaly GH levels are >2 ng/ml after an OGTT
What is the algorithm for acromegaly diagnosis (flow chart)?

How do we treat acromegaly?
- Microadenomas, macroadenomas that appear to be fully resectable and macroadenomas causing vision impairment transsphenoidal surgery is the treatment of choice
1. Macroadenomas abutting or adjacent to the chiasm should be decompressed surgically
2. Subsequent medical treatment may also be more effective after surgical debulking - For others, tx includes a long acting somatostatin analogue or pegvisomant (GH receptor antagonist)
What is the management strategy for pts with acromegaly (flow chart)?

A 44-year-old man is evaluated for a 2-year history of headache and 1-year history of diabetes mellitus and hypertension. His glove and shoe sizes have increased several times over the past 3 years, and he reports painful knees and hips. The patient also has sleep apnea and carpal tunnel syndrome. Medications are metformin and lisinopril.
On physical examination, blood pressure is 152/92 mm Hg, pulse rate is 82/min, and respiration rate is 16/min. Coarse facial features, frontal bossing, accentuated nasolabial folds, a large tongue, and thick hands and feet are noted.
What is the most appropriate diagnostic test?
- Serum IGF-1
- Don’t check GH because it is secreted in a pulsatile fashion (can be high or low depending on when you measure it)
What is prolactin?
- Polypeptide hormone produced mainly in ANT pituitary lactotrophs (some o/organs/tissues too)
- Tonically suppressed by hypothalamic dopamine from tuberoinfundibular cells, tuberohypophyseal dopaminergic system
- Induces, maintains lactation of the primed breast