Exam 2: Biochemistry Flashcards
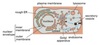
Cell
Compartmentalization
Topologically equivalent spaces:
Nucleus & Cytosol
Perinuclear cistern, ER cisterna, Golgi cisterna, Lysosomes, Transport vesicles & Endosomes
Movement between topologically inequivalent spaces requires translocators.
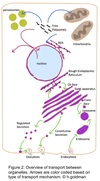
Cellular Transport
Mechanisms
- Gated transport
- Transmembrane transport
- Vesicular transport
Gated Transport
Large openings act as selective gates.
Between topographically equivalent spaces ⇒ does not cross a membrane.
Ex. nucleus ↔︎ cytosol
Transmembrane Transport
- Between topologically inequivalent spaces ⇒ crosses a membrane
- Uses translocators
- dependent on targeting signals
- protein moved in a denatured form
- Ex.
- import of nascent peptides into RER
- import from cytosol into mitochondria and peroxisomes
Vesicular Transport
- Between topologically equivalent spaces when each is membrane bound
- Transport vesicles carry proteins and membranes
-
Anterograde → “forward”
- ER to Golgi
-
Retrograde → “backward”
- Golgi → ER
- Endosomes → Golgi
- Ex.
- ER → Golgi
- Golgi → lysosomes / plasma membrane
- Endocytosis & Exocytosis

Vesicle Structure
- Inner layer formed from adaptor proteins links outer layer (cage) to the membrane
-
Cage proteins cover cytosolic surface forming a coat
- Assembly requires energy & GTP binding proteins
- Functions:
- collect specific membrane and soluble cargo
- direction formation of vesicles
- Removed before vesicles fuse
- Differs depending on destination and direction of movement
COPII Coating
Coats anterograde transport vesicles from ER → Golgi.

COPI Coating
Coats retrograde transport vesicles from the Golgi → ER.

Clathrin Coating
Transport vesicles from Golgi → endosomal compartments / plasma membrane.
Transport vesicles from plasma membrane → endosomes.

Vesicle Movement
Budding and targeting uses movement along cytoskeletal tracks:
Microtubules using kinesin and dynein
Actin filaments using Myosin II or Myosin V
Motor proteins recruited by Rab proteins.
Vesicle Targeting
Docking and fusion mediated by SNARE proteins.
SNARE proteins on transport vesicle bind complementary SNARE proteins on the target membrane.
Forces two membranes close together so lipid bilayers can fuse.

Genetic Code
Definition
The sequence relationship between the bases in the gene or mRNA and the amino acid in the protein.
3 consecutive nucleotides on mRNA ⇒ codon
Genetic Code
Characteristics
- 64 possible combinations of the 4 bases
- 61 code for AA
- 3 code for stop codons
- Codons are contiguous and do not overlap
- Degenerate ⇒ multiple codons can code for a single AA
- Unambiguous ⇒ each codon codes for one AA
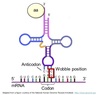
Wobble Hypothesis
The pairing between the codon and anticodon adheres to the usual base-pairing rules at the first two bases but is less strict for the third.
- First two bases predominate in tRNA selection
- Some tRNAs can bind to more than one codon that codes for the same AA
Start Codon
AUG ⇒ Methionine
N-terminal methionine formylated in prokaryotes ⇒ fMet
Stop Codons
UAA ⇒ U are awful
UAG ⇒ U are gross
UGA ⇒ U go away
Rules of
Protein Production
- Anticodon of tRNA pairs with codon of mRNA in anti-parallel fashion
- mRNA read in 5’ ⇒ 3’ direction
- Codons read sequentially by charged tRNAs
- Proceeds from N-terminus ⇒ C-terminus
Protein Synthesis
Stages
- Activation of amino acid
- Chain initiation
- Chain elongation
- Chain termination
- Co/post translational processing
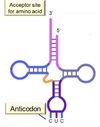
tRNA Structure
Acceptor end links the 5’ and 3’ ends forming clover structure.
3’-OH terminal CCA has AA attached to acceptor site.
Anticodon triplet base pairs with mRNA codon.
Amino Acid
Activation
“Charging the tRNA”
Catalyzed by aminoacyl-tRNA synthetase.
- 20-different aminoacyl tRNA synthetases recognizes ONE amino acid and ALL its cognate tRNAs
- Traps energy from hydrolysis of ATP → AMP + PPi in AA~AMP complex
- Two high energy bonds from ATP required
- Forms high energy bond between tRNA and AA used later to link AA to polypeptide chain
- Proofing mechanism to ensure correct AA attached
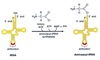
Ribosome Structure
Each subunit contains 3 critical sites:
A site ⇒ accepts the incoming aminoacylated tRNA
P site ⇒ holds tRNA and has ribosomal peptidyl transferase to form peptide bond
E site ⇒ temporarily holds deacylated tRNA until it exits the ribosome
Small Ribosomal Subunit
Functions
- Formation of the initiation complex
- Decodes the genetic information i.e. reads mRNA
- Binds both the 5’ end of mRNA and the tRNA-amino acid complex at the loading site
- Controls the fidelity of codon-anticodon pairing
Large Ribosomal Subunit
Functions
- Contains ribosomal peptidyl transferase activity that joins the AA to the polypeptide chain
- Contains translocation domain
- Contains tunnel where nascent peptide threaded
Translation Initiation

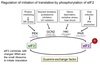
eIF2
Regulation
Activated by guanine nucleotide exchange factor.
Replaces GDP with GTP.
Inactivated by a GTPase.
Under conditions of stress, several kinases phosphorylate eIF2.
Phosphorylated eIF2 cannot bind gunine nucleotide exchange factor.
Low [ternary complex] ⇒ increased activation of amino synthesis genes.
mTOR
Mechanistic Target of Rapamycin
Serine/threonine kinase.
Major growth factor and nutrient sensor in the cell.
Able to modulate translational activity.
Activated via phosphorylation by insulin or IGF-1 via protein kinase B (Akt) pathway.
Also activated by nutrients, especially leucine.

eIF4 Regulation
-
Resources are plentiful
-
mTOR activated by protein kinase B (Akt) pathway
- Initiated by Insulin or IGF-1, leucine
- Activated mTOR phosphorylates 4E-BP1 (eIF4 binding protein)
- Phosphorylated 4E-BP1 releases eIF4E which can join the eIF4 complex
- Translation proceeds
-
mTOR activated by protein kinase B (Akt) pathway
-
Resources are scarce
- ↑ [AMP] ⇒ AMP kinase activation
- AMPK inhibits mTOR
- 4E-BP1 remains unphosphorylated
- eIF4E remains sequestered by 4E-BP1
- Translation is blocked

Translation Elongation
Depends on formation of peptide bond.
Aided by G-protein elongation factors (EFs).
Requires 2 ATP (charge tRNA) and 2 GTP (EFs) per cycle.
4-step repetitive process:
-
Charged tRNA brought into A site
- Aminoacyl-tRNA recruited by eEF1-GTP
- eEF1-GTP hydrolyzed by GTPase to eEF1-GDP and recycled.
- Amino acid attached to nascent polypeptide chain located in the P site by ribosomal peptidyl transferase
- Ribozyme component of large ribosomal subunit
- Ribosome advances 3 nucleotides towards 3’ end of mRNA ⇒ translocation
- Promoted by GTP-dependent eEF2
- Target for diphtheria toxin
- Promoted by GTP-dependent eEF2
- Empty tRNA moved to the E-site
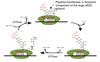
Translation Termination
- Release factors (RFs) recognize the stop codon and bind to the A site mimicking a tRNA
- Peptidyl transferase hydrolyzes the completed peptide chain from the final tRNA in the P site
- Hydrolysis of two GTP ⇒ GDP
- RFs use energy from GTP hydrolysis to induce conformation change in ribosome causing release of nascent polypeptide through exit tunnel
- mRNA released
- Large and small ribosomal subunits dissociate

Protein Processing
Overview
Co-translational vs Post-translational
Can occur in the cytoplasm or other cellular organelles.
- Protein folding
-
Covalent modifications
- Attachment of sugars, phosphate groups, or lipids
- Linked by disulfide bridge
- Cleavage
- Multiple subunits linked into quaternary structure

Protein Folding
Determined mostly by primary sequence of the peptide.
Primarily involves non-covalent interactions between AA side chains:
Hydrogen bonding
Van der Waals interactions
Electrostatic interactions
Native-like secondary structures assemble into domains ⇒ molten globules.
Aided by chaperones.
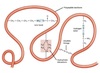
Chaperones
- Binds to “sticky” hydrophobic patches on nascent polypeptide chains.
- Prevents non-productive folding pathways or aggregation.
- Helps protein to fold by stabilizing intermediate conformations.
- Helps with creation of complexes.
- Stabilizes proteins as they move through intracellular organelles.
- Ex. Heat shock proteins
Disulfide Bond
Formation
Formed between the thiol groups of cysteine residues as proteins fold in the RER.
Catalyzed by protein disulfide isomerase.
Intramolecular disulfide bonds:
- Within a single polypeptide chain
- Usually stabilize the tertiary structure
Intermolecular disulfide bonds:
- Between seperate polypeptide chains
- Usually stabilize quarternary structure

Unfolded Protein Response (URP)
Mechanism
UPR normally inhibited when chaperone supply adequate.
“Extra” chaperones bind to sensing receptors in ER lumen.
When [misfolded proteins] ↑ or too many proteins made, “extra” chaperones dissociate from sensing receptors for use in the cell.
Sensing receptors become activated.
Unfolded protein response begins:
- ↓ protein synthesis
- ↑ chaperone production ⇒ ↑ capacity of ER to fold proteins
- Continued problem w/ refolding ⇒ aggregation/accumulation of abnormal proteins ⇒ destruction of unfolded proteins
- Failure to resolve high levels of unfolded proteins ⇒ apoptosis

Proteolytic Cleavage
Important step in the maturation processing of many proteins.
Proteases cleave by hydrolysis reaction.
-
Removal of N-terminus signal sequence
- Cleaved by signal peptide peptidase
- Associated wih the translocon
-
Removal of N-terminus initiator methionine
- Allows for addition of acetyl group or fatty acid chains
-
Cleavage of precursor proteins
- Activates zymogens

Insulin Processing
- On RER: Insulin synthesized as preproinsulin
- In ER: meti and signal sequence removed converting to proinsulin
- Proinsulin folds, stabilized by two interchain disulfide bonds
- In Golgi, proinsulin packaged into vesicles
- In vesicles: internal sequence removed (C peptide or connecting sequence)
- Regulated secretion of insulin and C peptide
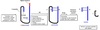
Trypsin
Functions of Glycosylation
- ↑ solubility to prevent agglutination
- Protects against proteolysis
- Helps folding and oligomerization
- Role in cellular sorting
- Recognition and antigenicity
N-linked Glycosylation
Carb attached to N-atom in side chain of asparagine.
Proteins mostly remain in ER or go to Golgi for export.
- asn-X-thr/ser motif
-
14 sugar residue transferred from dolichol phosphate ⇒ NH2 on Asn en bloc by protein-oligosaccharyl transferase
- On RER and associated w/ protein translocator
- Sugars added/removed as protein moves ER ⇒ Golgi
- Two categories w/ same 5 sugar core:
-
Complex oligosaccharides
- diverse sugars
-
High-mannose oligosaccharides
- mostly mannose
-
Complex oligosaccharides

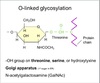
O-linked Glycosylation
Carb attached to O-atom in side chain of serine or threonine.
Much less common.
- Added in the Golgi
- GalNAc is the first sugar
- Added one at a time by glycosyltransferases
- Only a few residues
- Length varies
Cytosolic Glycosylation
- Proteins made by free ribosomes.
- Simpler modifications.
- Sugar moieties are not premade.
- Transferred as a precursor oligosaccharide.
Phosphorylation
Reversible post-translational +/- of phosphoryl groups.
Kinases ⇒ add
Phosphatases ⇒ remove
Serine/Threonine Kinases
Tyrosine Kinases
Protein Degradation
- Regulate 1/2 life
- Papidly degraded proteins usu. regulatory
- Transcription factors, signaling molecules, cytokines
- Papidly degraded proteins usu. regulatory
- Defective/damaged cells removed
- Ubiquitin-proteasome and lysosomal pathways
Ubiquitin-Proteasome Pathway
Cytosolic pathway
- Selective
- ATP dependent
- Quantitatively more significant
- Polyubiquitinated protein targeted to proteasome
- Ubiquitin ⇒ 76 AA polypeptide
Ubiquitination Steps
- E1 hydrolyzes ATP and adenylates ubiquitin
- Adenylated ubiquitin transferred to E2 via thioester bond
- E3 (Ubiqitin ligases) recognizes appropriate proteins and transfers ubiquitin
- ≥ 4 Ubiquitin added ⇒ proteasome degradation
N-end rule
“stabilizing” AA on N-terminal like Met ↑ 1/2 life
“destabilizing” AA on N-terminal like Arg ↓1/2 life
PEST Sequences
1+ internal PEST sequences target proteins for rapid degradation
Lysosomal Pathway
of
Protein Degradation
- Lysosomes have non-specific proteases called cathepsins
- Acid hydrolases
- Degrades mostly ingested proteins
- Role in cellular turnover
Cystic Fibrosis
- ∆F508 deletion
- Misfolding ⇒ ubiquitination ⇒ proteasomal degradation
Prokaryotic
Protein Synthesis
- Simpler with fewer factors
- Ribosomal subunits smaller
- Uses ATP to charge tRNA but not for protein synthesis
- No mRNA modifications or splicing
- Transcription & translation coupled
- mRNA polycistronic
- Uses purine-rich Shine-Dalgarno sequence in small ribosomal subunit to position mRNA, AUG, and tRNAiMet
- N-formylmethionine (fMet) is the first AA
- Recognized by body as foreign
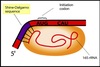
Prokarytote Translation
Initiation
Oriented using a Shine-Delgarnosequence
- Positions mRNA on small ribosome
- Purine-rich
- 6-10 bases upstream from initiating AUG
Streptomycin
(and related aminoglycosides)
High concentrations
- Binds S-12 of bacterial small ribosomal subunit
- Interferes with normal binding of fMet-tRNAiMet to P site
- Inhibits translation initiation
Low concentrations
- Causes misreading of mRNA
- Results in the wrong AA being inserted
Resistant strains of bacterial have altered ribosome that prevent drug binding.
Tetracycline
Binds to the small bacterial ribosomal subunit.
Interefers with binding of incoming aminoacyl-tRNA to the A-site.
Inhibits translation elongation.
Erythromycin
Interacts with large subunit of baterial ribosome.
Sterically hinders the exit tunnel.
Prevents release of nascent polypeptide.
In resistant strains, a single base of rRNA methylated and drug cannot bind.
Chloramphenicol
Inhibits peptidyl transferase activity.
Prevents transfer of growing peptide chain onto next AA residue.
Affects both prokaryotic and mitochondrial protein synthesis.
Reserved for severe cases of infection.
Diphtheria Toxin
Mechanism
- Toxin binds to plasma membrane and enters the cell.
- Catalyzes ADP ribosylation of EF-2 ⇒ inhibits translocation activity
- Inhibits elongation phase of protein synthesis

Co-Translational Targeting
Overview
All translation of nuclear genes begins on free ribosomes in cytosol.
Presence of ER-targeting signal on protein’s N-terminus targets peptide/ribosome pair to ER ⇒ becomes fixed.
Location of translation determines where protein will be ultimately trafficked and likely types of protein processing.
Signal Sequence / Leader Sequence
16-30 AA residue at N-terminus of protein
Has a hydrophobic core of 6-12 AA and one or more positively charged AA
Cleaved upon entry into the ER
Translocon
Water-filled channel in the ER membrane through which the nascent polypeptide chain passes.
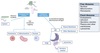
Translational ER Targeting
Mechanism
- Translation starts on free ribosome in cytosol
- Signal sequence on N-terminal emerges from ribosome
- Interacts with signal recognition particle (SRP)
- Temporarily stops translation ⇒ elongation arrest
- SRP targets ribosome-nascent chain complex to a docking protein on cytosolic face of RER
- Upon docking, GTP ⇒ GDP promotes insertion of nascent peptide chain into open translocon channel
-
SRP released
- Requires GTP hydrolysis
- Signal sequence removed by signal peptidase
- Elongation resumes
- When translation complete, ribosome released and translocon closes

Fates of Proteins
Synthesized on RER
- Resident ER proteins
- Formation of integral/transmembrane proteins
-
Default pathway
- No additional ER specific signals
- Moved to Golgi for ultimate constitutive secretion
- Additional tagging can target proteins to lysosomes or regulated secretory pathway
ER Resident Protein
Targeting
Possess retrieval signal sequences.
Allows proteins to be retrieved from Golgi and returned to the ER.
Most common is C-terminal KDEL (Lys-Asp-Glu-Leu)
Travel in COPI coated vesicles.
Ex. Disulfide isomerase
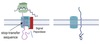
Intergral Membrane Protein
Targeting
- Assembled at the RER
- Majority are targeted and inserted into ER during synthesis
- Includes a stop transfer sequence
-
Transmembrane domain
- hydrophobic core
- can be a single domain or cross >20x
-
Translocon
- Can open the pore so polypeptide chain translocated into the ER lumen
- Can open laterally so hydrophobic regions enter the lipid bilayer
- Proteins can remain localized in ER membrane or transported in vesicles to other cellular membranes
- Default is the plasma membrane
Golgi Targeting
- Travel from ER ⇒ cis face of Golgi in COPII coated vesicles
- Move from cis ⇒ trans face to reach Trans-Golgi Network (TGN)
- Various modifications occur during transit
- Proteins sorted and placed into different vesicles in TGN
- Depends on tagged signal sequences
Golgi
Protein Modifications
-
Completion of N-linked glycosylation and O-linked glycosylation
- By glycosyl transferases
- Glycosaminoglycan chains added to core proteins ⇒ proteoglycans
Sorting in the Golgi
-
Mannose-6-phosphate (M6P) ⇒ lysosomes
- In clathrin coated vesicles
- Signals directing to secretory vesicles ⇒ regulated secretory pathway
- In specialized secretory cells
- Secreted and plasma membrane proteins selectively directed to apical or basolateral domains
- In polarized cells
- Requires specific signals
Secretory Pathways
Proteins segregated and concentrated into clathrin-coated vesicles within the trans-Golgi network (TGN).
Constitutive Pathway
- Default pathway
- Delivered from TGN ⇒ plsama membrane constitutively unless diverted elsewhere or retained in Golgi
- Operates in all cells
- Many soluble proteins
- Supplies plasma membrane with new components
Regulated Secretion Pathway
- Proteins diverted to secretory vesicles
- Concentrated and stored
- Extracellular signal stimulates secretion
- In specialized secretory cells only
- Small molecules can be actively transported from cytosol into preformed secretory vesicles via similar mechanism
- Ex. Histamine and neurotransmitters
- Often bound to specific macromolecules to store at high concentration without extra osmotic pressure
- Ex. Histamine ↔︎ Proteoglycans

Endocytic Membrane Transport
Pathway
Membrane-bound compartments inside cell.
Major sorting compartment.
Molecules transported from trans-Golgi membrane ⇒ endosomes in clathrin coated vesicles.
Classified as early, sorting, or late depending on stage post-internalization.
Endosomes can continue to develop into lysosomes or recycle back to Golgi.
Endocytotic vesicles can enter pathway ⇒ lysosomes or return to plasma membrane.
Lysosomal Targeting
- Protein undergo N-linked glycosylation in ER lumen
- Terminal mannose residue phosphorylated by phosphotransferase in Golgi ⇒ mannose-6-phosphate
- M6P recognized by specific Golgi receptors
- Receptor-protein complex buds off in clathrin-coated vesicle
- Vesicle fuses with endosomes
- Low pH ⇒ dissociation of glycoprotein from M6P receptor
- Vesicle w/ fully processed lysosomal enzymes bud off from endosomes & fuse with lysosomes
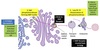
I-Cell Disease
AR lysosomal storage disease
Deficient phosphotransferase ⇒ no M6P tag
Material cannot be broken down ⇒ inclusions
Free Ribosome
Products
Proteins made by cytosolic ribosomes:
Retained in the cytosol (default pathway)
Targeted to nucelus, mitochondria, or peroxisomes by specific signals
Nuclear Localization Signal
(NLS)
Short stretch of basic amino acid residues.
Ex. Lysine or Arginine
Integral to the protein ⇒ not cleaved.
Usually sequestered until transport required ⇒ conformational change exposes NLS
Nuclear Import
Mechanism
- Protein translated by free ribosomes
- Nuclear localization signal bound by importin
- Protein-importin complex docks and translocated through nuclear pore complex
- Inside the nucleus, protein released facilitated by Ran-GTP
- Ran-GTP carries importin back to cytosol
- Hydrolysis of GTP releases importin
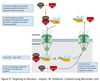
Nuclear Export
Mechanism
- Nuclear export signals (NES) binds to exportin and Ran-GTP
- Trimeric complex transported through nuclear pore
- Hydrolysis of GTP in cytosol dissociates complex
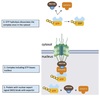
Nuclear Transport
Driving Force
Transport through nuclear pore driven by concentration gradient of Ran-GDP in cytosol and Ran-GTP in nucleus.
Swyer Syndrome
Due to mutations in the NLS of SRY protein.
SRY unable to enter nucleus ⇒ no testis-developmental signals
XY individuals born as phenotypic females
Mitochondrial-Targeting Signal
(Matrix-targeting Signal)
Amphiphatic alpha helix
- Basic residues on one side
- Hydrophobic residues on the other
One or more signal sequences direct protein to the co rrect mitochondrial sub-compartment.
Mitochondrial Matrix Targeting
Mechanism
- Protein translated by free ribosomes
-
Chaperones bind nascent protein maintaining in unfolded state
- Requires ATP
- Matrix-targeting signal sequence on N-end recognized by translocase in the outer membrane (TOM complex)
- N-terminus moved into intermembrane space
- Signal sequence binds translocase in the inner membrane (TIM complex)
- Protein moved into the matrix
- Protein spans both matrices for a short time
- Signal sequence removed by signal peptidase in the matrix
-
Mature protein forms within the matrix with help of chaperones
- Requires ATP
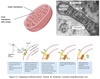
Mitochondrial Subcompartment
Targeting
Internal signals, rather than N-terminal.
Some with stop-transfer signals.
Can target proteins to outer membrane, inner membrane, and inner membrane space.
Peroxisomal Targeting
Peroxisomal proteins selectively imported from the cytosol via transmembrane tansport.
Most with a peroxisomal targeting sequence (PTS) at the C-terminus⇒ 3-amino acid (Ser-Lys-Leu)
Some with target sequence at the N-terminus.
Import requires peroxins
Acts as both chaperones and receptor.

Zellweger’s Syndrome
AR disorder
Inability to correctly target proteins to matrix of peroxisomes.
Neurological impairment leads to death at an early age.
Protein Targeting
Summary

Mechanisms of Endocytosis
- Pinocytosis
- Receptor-mediated Endocytosis
- Phagocytosis
Pinocytosis
Cell ingests fluids, molecules, and particles.
Performed in virtually every cell type.
Contitutive and nonspecific.
Vesicles do not contain a clathrin coat.
Process:
- Substances to be taken in contacts extracellular surface of plasma membrane.
- Surface becomes indented.
- Small pinocytoic vesicles dynamically form in the plasma membrane.

Receptor-mediated Endocytosis
Mechanism
- Cargo protein binds specific receptors forming a clathrin-coated pit
- Pit buds from plasma membane forming a coated vesicle
- Vesicles lose their coat within the cell and fuse with early endosomes
- Early endosomes are the main sorting station
- Endosomes mature and pH ↓
- Patches of membrane invaginate into lumen forming intralumenal vesicles ⇒ multivesciular bodies
- Ligands dissociate from receptors
- Most receptors recycled to plasma membrane via transport vesicles
- Ligands digested in lysosomes.

Transcytosis
Sometimes receptors/ligands can be transcytosed to another part of the plasma membrane.
Ex. epithelial cells in lamina propria move Ab from basolateral domain to apical domain.

Low-density Lipoprotein (LDL)
Transport
Ex. of receptor mediated endocytosis
- LDL binds LDL receptor on cell surface
- LDL/LDL receptor internalized in clathrin-coated pits ⇒ clathrin-coated vesicles
- Vesicles lose coat and fuse with early endosomes
- Low pH in endosome ⇒ dissociation of LDL from receptor
- Continue through endosomal pathway to lysosomes
- LDL hydrolyzed to free cholesterol
Phagocytosis
- Phagocytic cell engulfs pathogen in phagosome
- Phagosome fuses with lysosome ⇒ phagolysosome
- TACO (tryptophanaspartate-containing coat protein) coat must be removed before fusion
- Internalized membrane components recycled
- Pathogen digested in lysosomes
Process called heterophagy.

Mycobacteria
ex. TB bacillus
Avoids digestion by preventing TACO coat from being removed.
Can sometimes be killed via autophagy.
Listeria monocytogens
Can escape from phagosomes.
Bacteria secretes a protein that destroys phagosome membrane.
Autophagy
Overview
Degradation pathway for cellular proteins and organelles.
3 general types:
- Macroautophagy
- Microautophagy
- Chaperone-mediated autophagy

Macroautophagy
- portions of cytoplasm or whole organelles surrounded by a vacuole
- forms a double-membraned sac ⇒ autophagosome
- outer membrane fuses with lysosome ⇒ autolysosome
- inner membrane and contents digested

Microautophagy
Non-specific cytoplasmic proteins enter via invagination of lysosomal membrane.

Chaperone-mediated autophagy
- Specific proteins with targeting signals directed into lysosomes
- Aided by heat-shock chaperone proteins
- Requires specific receptors on lysosomal surface
- Significant protein degradation mechanism in liver and kidney
- Activated during starvation

Neimann-Pick Disease
Dysfunctional metabolism of sphingolipids.
Due to defects in autophagy.
Accumulation of large amounts of cholesterol and lipids in lysosomes.
Crinophagy
Disposal of excess proteins stored in secretory vacuoles by fusion with lysosomes.
Rare process occuring in few cell types.
Happens when it will likely a long time before products needed again.
Ex. mammotrophs of anterior pituitary & prolactin.
Fibril Forming
Collagens
Forms rope-like structure
Characteristic banding pattern
High tensile strength
Type I, II, and III collagens
Type I Collagen
Fibril forming
90% of body collagen
Found in:
Dermis
Tendons / Ligaments
Bone
Fascia
Organ capsules
Cornea

Type II Collagen

Type III Collagen

Networking Forming
Collagens
Forms a 3-D mesh
Ex.
Type IV Collagen ⇒ anchoring plaques in basal lamina
which connects to
Type VII Collagen ⇒ anchoring fibrils of lamina reticularis

Fibril-Associated
Collagens
Flexible collagens with interrupted helices.
Bind to fibrils and connects then to ECM.
Collagen types IX and XII
Type I Collagen
Structure
Triple helix formed of 2 identical α1 chains and an α2 chain.
- Rich in proline and glycine
- Proline ⇒ kinks peptide chain for helix formation
- Glycine ⇒ allows tight turns
- Select prolines and lysines hydroxylated
- Site for stabilizing interchain H-bonds
- 3-residue repeating motifs
- Gly-Pro-X
- Gly-X-Hyp
α chains ⇒ triple helix (procollagen) ⇒ tropocollagen ⇒ fibrils ⇒ fibers ⇒ fascicles ⇒ tendons/ligaments
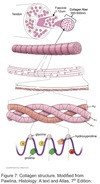
Type I Collagen
Synthesis
-
COL genes transcribed ⇒ α1 and α2 chains
- mRNA processed
- Moved into cytosol via gated transport
- mRNA translated by free ribosomes ⇒ makes preproprotein
- Signal sequence ↔︎ signal recognition particle ⇒⇒⇒ RER
- Nascent polypeptide enters translocon ⇒⇒⇒ RER lumen
- Signal peptide cleaved by signal peptidase
- Converts preproprotein ⇒ proprotein
- Select proline and lysine residues hydroxylated by two ascorbate dependent enzymes
- Pro ⇒ hydroxyproline by proline hydroxylase
- Lys ⇒ hydroxylysine by lysyl hydroxylase
- Select hydroxy lysines receive O-linked glycosylation with galactose or glucose
-
Triple helix forms from C-terminus to N-terminus ⇒ procollagen
- Stabilized by intra- and interchain hydrogen and disulfide bonds
- Aided by chaperones
- Procollagen packaged into secretory vessicles
- Constitutively secreted via default pathway
- In ECM, hydrophilic N and C-terminal propeptides cleaved by procollagen peptidases ⇒ forms tropocollagen
- Tropocollagen aggregate into fibrils d/t hydrophobic effect
- Fibrils stabilized by lysyl oxidase
- Copper-requiring enzyme
- Cross-links the fibrils via staggered covalent bonds
- Fibrils stabilized by lysyl oxidase
- Fibrils aggregate to form mature collagen fibers
Scurvy
Dietary Vit C deficiency
Impaired function of proline and lysyl hydroxylases
-
Initial symptoms:
- Fatigue, malaise, gum inflammation
-
Progressive symptoms:
- Depression
- Swollen and bleeding gums
- Loosening or loss of teeth
- Eecchymoses
-
Secondary iron deficiency anemia
- Due to blood loss and ↓ nonheme iron absorption
- ↑ risk with malabsorption disorders, cancers, end-stange renal disease

Osteogenesis Imperfecta
(OI)
90% of cases due to mutations in COL1A1 or COL1A2 genes.
Results in abnormal collagen structure.
-
Type I OI
- Mildest form
- Due to mutation resulting in abnormal protein that does not leave ER or form procollagen
- nonsense or frameshift
-
Types II-IV OI
- More severe
- Caused by missense mutation replacing glycine residues
- Alpha-chains cannot form tight turns
- Steric hinderance and bulge in triple helix during procollagen formation
- Protein degraded

Ehlers-Danlos Syndrome
(EDS)
Due to mutations in either collagen genes or mutations in proline/lysyl hydroxylases.
- Joint hypermobility
- Skin hyperextendibility
- Atrophic scar formation
- Arterial, intestinal, and/or uterine fragility

Menkes Syndrome
Mutation in gene critical for regulating copper level.
X-linked recessive disorder
- Copper accumulates in kidney and intestines
- Inadequate Cu in other tissues, esp brain
- Impact function of copper-containing enzymes
- Lysyl oxidase involved cross-linking of collagen fibrils
- Sx.
- Sparse, brittle, and twisted hair
- Failure to thrive
- Lack of muscle tone
- Seizures
- Progressive brain deterioration
- Often do not live past 3 y/o

Intermediary Metabolism
All the chemical changes that are involved in the occurance and continuance of life.
Ability to accomplish these changes at constant body temperature requires enzymatic catalysis & thermodynamic coupling of endergonic and exergonic processes.
Pathway
The series of steps involved in the breakdown or synthesis of major biological constituents.
Cycle = pathway that regenerates the initial substrate
Catabolic and anabolic pathways linked via ATP.
Catabolism
Degradative (complex to simpler)
Oxidative
Exergonic
ATP generating
Often requires NAD+ or FAD
Anabolism
Sum of the pathways that are involved in synthesis and growth.
Reductive
Energy consuming
ATP utilization
Often requires NADPH
Metabolic rate
Expression of enthalpic change.
Gives the normalized total heat production per unit time.
Basal metabolic rate
Measured in a resting state (awake laying still)
Energy requirements for:
- involuntary muscle work
- maintenance of osmotic gradients
- maintenance of body temperature
- turnover and synthesis of cell constiuents
Gibb’s free energy
(ΔG)
ΔG = ΔH - TΔS
H= enthalpy S= entropy T= temp in kelvins
The energy change occuring under conditions of constant pressure and temperature.
Additive function and independent of pathway.
Total ΔG of a process can be expressed as the sum of ΔG changes of individual steps.
Thermodynamic Coupling
Coupling of an exergonic and endergonic reaction so that net ΔG is negative and reaction can occur.
A common intermediate must exist.
In an enzyme catalyzed reaction, the common intermediate may not be free and may only exist on the enzyme.
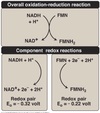
Standard reduction potentials
(Eº)
The relative affinity of a molecule, atom, or ion for electrons taken under standard conditions where reactants and products are at unit activity (~1 M) then compared to the proton/hydrogen electrode (H+ and ½ H2)
Often expressed as values corrected to pH 7 (Eº’)
Interpreted as the relative affinity of the system for electrons as compared to that of a proton:
- Negative reduction potential indicates a weaker affinity for electrons than a proton.
- Positive reduction potential indicates a stronger affinity.
Cellular reduction potential
Dependent on the ratio of oxidant to reductant agent concentration in a cell.
Nernst equation
E = Eº’ + 2.3 RT/nF log [oxidant] / [reductant]
R = gas constant
F = Faraday constant
Relationship of reduction potential to ratio of oxidant to reductant agents in a cell.
High energy bonds
Describes a bond which has a large negative standard Gibb’s free energy (ΔGº’) of hydrolysis
Minimum of -7.0 kcal/mol
Indicated with a ~
Adenosine triphosphate
(ATP)
- Contains two phosphoanhydride bonds which have a ΔGº’ of hydrolysis of approximately -7.3 kcal/mol each.
- Biological utilization of these high energy bonds require the ATP form.
- One or both bonds may be used in a reaction.
- Phosphoryl group(s) of ATP can be transferred to acceptor molecules to generate activated intermediates for metabolism.
- Can function as coenzyme-cosubstrates.
Pyrophosphate
(PPi)
Use of both phosphoanhydride bonds of ATP is the functional equivalent of using 2 ATP’s.
Reactions coupled to the hydrolysis of the pyrophosphate (PPi) product to 2 orthophosphates (Pi) by pyrophosphatase which drives reactions forward.
Adenylate kinase
Catalyzes the reversible reaction:
AMP + ATP ⇔ 2 ADP
Adenine nucleotide cosubstrate metabolic pool
Total concentration of adenine nucleotide pool essentially constant, however, ratio of adenine nucleotides vary with metabolic state of the cell.
Concentrations of ATP, ADP, and AMP in a cell are in rapid equilibrium due to activity of adenylate kinase.
The cosubstrate pool communicates between and significantly influences the different pathways of the cell which utilizes those cosubstrates.
AMP level most sensitive parameter to change in pool and usually initiates the responses to decreased ATP levels.
Energy charge
Energy charge = ½ • ( [ADP] + 2[ATP] ) / ( [AMP] + [ADP] + [ATP])
A stoichiometric expression of the mold fraction of high energy phosphate bonds present relative to the maximal high energy bonds possible.
0 = nucleotides are totally in the form of AMP
1 = nucleotides are totally in the form of ATP
ΔG for ATP hydrolysis
ΔGATP→ADP = ΔGº’ATP→ADP + RT ln ( [ADP] x [Pi] / [ATP] )
Actual ΔG for ATP hydrolysis in a cell is dependent upon and may be calculated from the concentrations of ATP, ADP, and inorganic phosphate.
May be very different from the ΔGº’.
In a resting cell may be as high as = 15 kcal/mol which is equivalent to an energy charge of approximately 0.9.
“High energy charge”, “large negative ΔG of ATP hydrolysis”, and “resting cell” all indicate that ATP is plentiful.
Pathway regulation
- Pathways for synthesis and breakdown of the same constituent are never the same, although individual steps may be reversible and utilized in both pathways.
- Opposing pathways typically occur in seperate cellular compartents and/or different tissues or organs.
- Regulation of pathway enzyme activity by:
- product inhibition
- allosteric regulation
- covalent regulation i.e. phosphorylation
- Changes in the rate of synthesis/degradation of an enzyme or its mRNA.
Glucose Uptake
- Glucose transported across cell membranes by:
- Facilitated diffusion via GLUT transports down their concentration gradients.
- In renal and intestinal epithelium, transported against its concentration gradient by Na+-glucose co-transporters (SGLTs)
GLUT transporters
- Glucose-dependent tissues such as RBC’s and brain have low Km insulin-independent GLUT1 or GLUT3 transports respectively.
- In peripheral tissues such as muscle which are glucose-independent, GLUT4 transports have a low KM but is insulin-dependent ⇒ allows cross regulation.
- The liver which does not rely on glucose for energy, uses the GLUT2 transporters have a high KM for glucose but is insulin-independent.
- Limits glucose uptake to conditions when blood glucose levels are high.
- Allows the transporter to act as a sensor of high blood glucose levels.
* Normal fasting blood glucose levels are 3.9 - 5.5 mmol/L.

GLUT 4 Regulation
- Located in muscle and adipose tissue.
- Relatively low Km for glucose so would transport around normal fasting levels.
-
Insulin-dependent glucose transporter:
- When insulin is absent, the transporters are removed from the plasma membrane and sequestered into vesicles.
- Functional but not in the membrane.
- Insulin signaling stimulates the movement of the transporter from internal stores to the plasma membrane.
- When insulin is absent, the transporters are removed from the plasma membrane and sequestered into vesicles.
-
In skeletal muscle, exercise stimulates GLUT4 translocation to the plasma membrane through AMP-activated protein kinase (AMPK) via unknown mechanism.
- Long-term exercise also increases the amount of GLUT4 in the muscle cell.

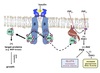
Mechanism of Insulin
GLUT4 Activation
- Binding of insulin to the α-subunit of its receptor activates a tyrosine kinase domain resulting in auto-cross-phosphorylation of tyrosine residues in the β-subunits.
- Negative charge of the phosphates causes IRS (insulin receptor substrate) proteins to bind to the β-subunit.
- IRS proteins phosphorylated at two Tyr residues by the kinase activity of activated insulin receptor.
- Phosphorylated-IRS dissociate from the receptor then bind to and activate proteins with SH2 domains i.e. PI-3-kinase (Phosphatidylinositol-3-kinase).
- PI-3-kinase phosphorylates PIP2 to PIP3.
- PIP3 activates PDK-1 (phosphoinositide-dependent kinase).
- PDK-1 activates downstream effectors Akt and PKB which results in the movement of GLUT4 to the cell surface in adipose and muscle, increasing glucose uptake.
Akt/PKB also:
- phosphorylates and inactivates GSK3 (glycocen synthase kinase 3) resulting in increased glycogenesis.
- Activate amino acid uptake and protein synthesis
- Increase lipid synthesis
- Inhibit gluconeogenesis
- decrease cAMP levels by activating phosphodiesterase
- various gene expression modulations both +/-
- Increases protein synthesis by activating a kinase (mTOR) that ultimately results in the activation of eIF4 and EF2 from protein translation.
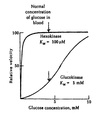
Glucose Phosphorylation
- Phosphorylation of glucose prevents back diffuse out of the cell via the transporter and commits it for use in that cell.
-
Hexokinases
- Found in tissues such as muscle and brain.
- Has a low KM for glucose
- Can phosphorylate other monosaccharides but affinity for glucose considerably higher
- Show product inhibition by glucose-6-phosphate.
-
Glucokinase
- Found in liver and pancreatic β-cells
- Has a high KM for glucose
- Shows no direct product inhibition

Glucokinase Kinetics
- Despite being monomeric, glucokinase displays sigmoidal kinetics towards glucose.
- The inflection point of the glucokinase enzyme curve is such that small changes in blood glucose levels causes significant changes in enzymatic activity.
- When blood glucose levels are high the hepatic glucokinase becomes significantly more active.
- Hexokinase, however, is fully saturated a normal concentrations of blood glucose.
Equation for glycolysis
glucose + 2 NAD+ + 2 ADP + 2 Pi
→
2 pyruvate + 2 NADH + 2 ATP + 2 H2O + 2 H+
General stages of glycosis
- Priming steps
- Two ATP-linked phosphorylations
- Functions to prevent diffusion of the intermediates of the pathway out of the cell.
- Oxidation/reduction with the production of NADH
* Results in the generation of ATP - Re-oxidation of the NADH produced.
Substrate-level phosphorylation
The synthesis of ATP involving two coupled reactions linked by a common intermediate containing a high-energy bond.
Steps of glycolysis
- Irreversible phosphorylation of glucose at carbon-6 by hexokinase using ATP•Mg2+ to produce glucose-6-phosphate.
- Hepatic isozyme is glucokinase.
- Traps glucose because cell membrane is impermeable to phosphate esters.
- Commits glucose to intracellular metabolism but NOT glycolysis.
- Freely reversible aldose-ketose isomerization of glucose-6-phosphate to fructose-6-phosphate by phosphoglucose isomerase.
- Irreversible phosphorylation of fructose-6-phosphate to fructose-1,6-bisphosphate using ATP by phosphofructokinase (PFK1).
- True commitment step and primary regulatory site for glycolysis.
-
Fructose-1,6-bisphosphate split into two triose phosphates: dihydroxyacetone phosphate (DHAP) and glyceraldehyde-3-phosphate (GAP) by aldolase.
- DHAP freely interconverted to GAP by triose phosphate isomerase.
- Only GAP enters next stage of glycolysis.
-
Glyceraldehyde-3-phosphate dehydrogenase catalyzes the reversible oxidation of glyceraldehyde-3-phosphate to 1,3-bisphosphoglycerate.
- NAD+ converted to NADH **
- Phosphate from the carboxyl group from 1,3-bisphosphoglycerate is reversibly transferred to ADP to produce ATP and 3-phosphoglycerate by 3-phosphoglycerate kinase.
- First substrate-level phosphorylation.
- Additional mutase found in erythrocytes which transfers carbonyl phosphate of 1,3-bisphosphoglycerate to carbon-2 to produce 2,3-bisphosphoglycerate (2,3-BPG).
- 3-phosphoglycerate converted to 2-phosphoglycerate by phosphoglycerate mutase.
- 2-phosphoglycerate converted to phosphoenolpyruvate (PEP) by enolase with loss of H2O.
- Phosphate of PEP is transferred to ADP by pyruvate kinase (PK) to produce pyruvate and ATP.
- Irreversible
- Has a ΔGº’ of -14 kcal/mol
- NADH produced is recycled back to NAD+
- In aerobic conditions via malate shuttle.
- In anerobic conditions pyruvate converted to lactate via lactate dehydrogenase.

PFK1
Importance
Most important regulatory step in glycolysis because:
- It is the commitment step for the glycolytic pathway.
- It is allosterically modulated by many metabolic intermediates and products.

PFK1
Allosteric Regulation
Allosteric effectors act by influencing the equilibrium between the active and inhibited forms of the enzyme.
Binds preferentially to one form or the other and stabilizes that form.
-
Activators ⇒ high energy signals
- Citrate
- ATP
- Fructose-2,6-Bisphosphate***
-
Inhibitors ⇒ low energy signals
- ADP
- AMP

Role of
Fructose-2,6-Bisphosphate
Fructose-2,6-bisphosphate (F-2,6-P2) is a positive heterotropic effector of PFK1.
F-2,6-P2 is synthesized and degraded by a multifunctional enzyme with both:
- kinase activity ⇒ PFK2
- phosphatase activity ⇒ F-2,6-Bisphosphatase

PFK2
Isozymes & Regulation
Multifunctional enzyme reponsible for synthesis and degradation of F26BP.
PFK2 & F26BPase
Phosphorylation of either domain inhibits its catalytic activity.
Liver:
PFK2 enzyme a substrate for cAMP-dependent PKA.
Phosphorylation site for liver PFK2 isozyme lies within the kinase domain.
Glucagon ⇒ phosphorylation ⇒ ↓ kinase activity & ↑ phosphatase activity ⇒ ↓ [F26BP] ⇒ ↓ PFK-1 activity
Skeletal Muscle:
PFK2/F26BPase isozyme has no phosphorylation sites.
Is not covalently regulated.

Hepatic PFK1 Regulation
- Hepatic PFK1 is primarily regulated by the [F-2,6-BP]
- The primary function of F-2,6-BP in the liver is to make PFK1 sensitive to regulation by glucagon and other hormones.
- The liver does not consume glucose as fuel during times of need – rather it makes glucose for use by other tissues.

Pyruvate Kinase
Regulation
Covalent Regulation
(Liver Only)
-
Fasted state:
- Glucagon → cAMP → PKA → phosphorylated PK ⇒ PK inhibited
-
High glucose:
- Insulin → ↑ phosphatase activity → dephosphorylation of PK ⇒ PK activation
Allosteric Regulation
(All cells)
-
Fasted state:
- Acetyl CoA
- Long chain fatty acid
- Alanine
-
High energy state:
- ATP
- F-1,6-BP

Fermentation
Glycolysis occurring under anaerobic conditions.
Lactate Fermentation
Pyruvate + NADH → lactate + NAD+
Catalyzed by lactate dehydrogenase.
Occurs in mammalian tissues specifically skeletal muscle.
Allows glycolysis to continue under anerobic conditions.
Lactate enters the blood and is ultimately reconverted to glucose in the liver via gluconeogenesis.
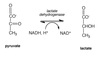
Alcoholic fermentation
Occurs in microorganisms such as yeast.
1. Pyruvate → CO2 + acetaldehyde
Catalyzed by pyruvate decarboxylase.
2. Acetaldehyde + NADH → ethanol + NAD<strong>+</strong>
Catalyzed by alcohol dehydrogenase.
Malate-Aspartate Shuttle
- Electrons are transferred from cytosolic NADH to oxaloacetate forming malate and NAD+.
- Malate enters the mitochondrial inner membrane via malate/α-ketoglutarate transporter.
- Inside the matrix, malate is reoxidized by malate dehydrogenase and NAD+ to form OAA and NADH.
- OAA is converted to aspartate via a transamination reaction.
- Aspartate is transported back to the cytosol via a glutamate/aspartate transporter.
- In the cytosol the aspartate undergoes transamination to reform OAA.
*Shuttle is readily reversible: important in gluconeogenesis.

Glycerol-3-Phosphate Shuttle
Couples the cytosolic oxidation of NADH with the mitochondrial reduction of FAD.
- Cytoplasmic NADH utilized by glycerol-3-phosphate dehydrogenase to convert dihydroxyacetone phosphate to glycerol-3-phosphate.
- Glycerol-3-phosphate is then converted back to DHAP by mitochondrial version of the dehydrogenase which resides on the inner mitochondrial membrane. FAD is reduced to FADH2.
- Electrons from FADH2 are trasferred to the electron carrier Q which enters the respiratory chain as QH2.
Pentose Phosphate Pathway
Basics
- Alternate pathway from glucose-6-phosphate
- Primary function is generation of NADPH
- Important in neutralization of ROS
- Used to support fatty acid biosynthesis (cytosol)
- Serves as a source for ribose-5-phosphate synthesis
- Used in synthesis of nucleic acids
- Used to produce glyceraldehyde-3-phosphate
- Fed into TCA and ETC cycles
- Pathway can be divided into two sections:
- Oxidative
- Non-oxidative
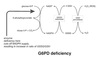
Glucose-6-Phosphate Dehydrogenase
(G6PD)
Rate-limiting and regulated step in PPP
Stimulated by G6P and NADP+
NADPH is a competitive inhibitor
Glucose-6-Phosphate Dehydrogenase
(G6PD)
Deficiency
- Most common of all enzyme deficiency-related diseases
- X-linked
- Cuts off cell’s supply of NADPH
- Affects RBC’s most who cannot produce new proteins
- Oxidative stress leads to hemolysis and anemia
- Infection most common
- Drugs, chemicals, certain foods
- Fava beans
- Anti-malarials
- Certain antibiotics
- Naphthalene
- Heterozygotes have some resistance against malaria
- Therefore gene prevalent in the Mediterranean and Africa
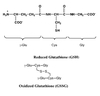
Glutathione
- Glutathione and the glutathione peroxidase system is the principal antioxidant defense system in mammalian cells
- Glutathione is a tripeptide which contains a central Cys residue.
- Reduced form (GSH)
- Oxidized form (GSSG)
Metabolism of Other Monosaccharides
- Glucose preferred but cells will also use other sugars
- Fructose and galactoase are significant in the diet
- Mannose important components of glycoproteins
- Metabolism of other sugars are fed into glycolytic pathways
- Sugars must be phosphorylated by the cell before they can be used.
- Hexokinase/glucokinase can phosphorylate other monnosaccharides but their KM are significantly higher than glucose’s
- Galactokinase present in most cells
- Fructokinase in liver only

Fructose Metabolism
-
Fructokinase (found primarily in the liver) phosphorylates fructose at the 1 position producting fructose-1-phosphate
- No mechanism exists to convert F-1-P to G-1-P
- Aldolase B (liver isozyme of aldolase) can split both F-1-P and F-1,6-P2 to form dihydroxyacetone phosphate (DHAP) and glyceraldehyde.
-
Glyceraldehyde formed can be:
- Phosphorylated by triose kinase to form glyceraldehyde-3-phosphate → enter glycolysis
- Reduced to glycerol by alcohol dehydrogenase then phosphorylated by glycerol kinase to form glycerol-3-phosphate which is then converted to DHAP by glycerol phosphate dehydrogenase.
- DHAP converted to GAP by isomerase.

Essential Fructosuria
- Due to deficiency of fructokinase
- Relatively benign
- Leads to accumulation of fructose in the urine
Hereditary Fructose Intolerance
(HFI)
- Autosomal recessive
- Absence of aldolase B (liver isoform)
- Results in accumulation of fructose-1-P in the liver
- Depletes levels of ATP and Pi
- Low Pi levels inhibit glycogenolysis
- Low ATP levels inhibit gluconeogenesis
- Low Pi activates AMP deaminase in muscle
- Results in increased purine catabolism and hyperuricemis → gout
- Low Pi prevents phosphorylation of ADP so adenylate kinase will convert 2 ADP → ATP + AMP
- AMP degraded to urate
- Symptoms include:
- Vomiting
- Hypoglycemia
- Jaundice
- Metabolic acidosis
- Coma
Galactose Metabolism
Most tissues are cabable of metabolizing galactose.
- Galactokinase phosphorylates galactose at the 1 position producing galactose-1-P.
- Galactose-1-phosphate uridylyltransferase (GALT) switches galactose for glucose from UDP-glucose producing UDP-galactose.
- UDP-galactose can interconverted to UDP-glucose by UDP-glucose-4-epimerase.

Uridylyltransferase (GALT) deficiency
- Most common cause of galactosemia
- Symptoms include:
- Failure to thrive
- Liver damage
- Bleeding
- Sepsis
- Cataracts - later on
- If galactose restricted diet provided within the first 10 days the most severe complications can be avoided:
- Neonatal death
- Liver failure
- Intellectual disability
- Children with galactosemia remain at risk for developmental delays and problems with speech and mother function.
Galactokinase Deficiency
- Rare disorder
- Morbidity limited to cataract formation
Coenzyme A
(CoA)
- Derived from ATP and pantothenic acid (Vit B5)
- Contains a reactive thiol group (-SH) that is covalently linked to the acetyl group via a thioester bond
- Acetyl~CoA readily donates acetyl groups to other acceptors
Pyruvate dehydrogenase complex
(PDH)
- Member of the α-ketoacid dehydrogenase family
- Large multienzyme complex which contains 3 enzymes in multiple copies
- Very efficient due to close spatial proximity of complexes
Enzyme subunits
- E1: pyruvate dehydrogenase (aka pyruvate decarboxylase) #30
-
E2: dihydrolipoyl transacetylase #60
- Acts as a swinging arm between E1 & CoA preventing substrate from diffusing away ⇒ substrate channeling
- E3: dihydrolipoyl dehydrogenase #12
- Contains two stoichiometric coenzymes/cosubstrates
- NAD
- CoA
- Contains three catalytic coenzyme prosthetic groups
- thiamine pyrophosphate (TPP)
- FAD
- lipoic acid
- Contains two regulatory enzymes associated with but not part of the complex
- PDH kinase
- PDH phosphatase

Steps for oxidative decarboxylation of pyruvate
- Pyruvate is decarboxylated and the acetyl group is attatched to thiamine pyrophosphate (TPP) coenzyme of pyruvate decarboxylase (E1).
- Acetyl group transferred to the lipoic acid covalently bound to dihydrolipoyl transacetylase (E2),
- The acetyl group, bound as a thioester to the side chain of lipoic acid, is transferred to free CoA.
- The sulfhydryl form of lipoic acid is oxidized by FAD-dependent dihydrolipoyl dehydrogenase (E3) to regenerate the oxidized lipoic acid.
- FADH2 on E3 is reoxidized to FAD as NAD+ is reduced to NADH2 + H+.

PDH complex
Regulation
Allosteric regulation
-
Inhibitors (feedback inhibition)
- Acetyl~CoA
- NADH
Covalent regulation
(main mechanism)
- PDH inactivated by phosphorylation by PDH kinase
- PDH activated by dephosphorylation by PDH phosphatase
PDH kinase and PDH phoshatase are in turn allosterically regulated:
-
PDH kinase
-
activated (high-energy signals)
- ATP
- acetyl-CoA
- NADH
-
inhibited
- pyruvate
-
activated (high-energy signals)
-
PDH phosphatase
- activated
- ↑ [Ca2+]
- important in skeletal muscle
- ↑ [Ca2+]
- activated

Niacin and Thiamine Deficiency
Niacin (B3) → NAD+/NADH
Thiamine (B1) → TPP
Lack of either will reduce/block activity of PDH complex.
↓ ATP synthesis
CNS dependent on ATP ⇒ reduced CNS function
Arsenic
Poisoning
Arsenic in the form of trivalent arsenite (AsO33-) forms a stable complex with thiol groups of lipoic acid.
Lipoic acid unable to serve as coenzyme for PDH.
TCA cycle
General
Acetyl~CoA + 3 NAD+ + FAD → 2 CO2 + 3 NADH + FADH2
- 8 steps
- 4 oxidations that produce NADH or FADH2
- one substrate-level phosphorylation producing GTP
- Except for succinate dehydrogenase which is embedded in the inner mitochondrial membrane all enzymes are located in the matrix
Steps of the TCA cycle
-
Formation of citrate.
- Condensation of acetyl~CoA and oxaloacetate (OAA) catalyzed by citrate synthase.
- Reaction made irreversible by the hydrolysis of the thioester bond of acetyl~CoA.
- Facilitated by an enzyme-bound intermediate, citroyl~CoA
- Citrate synthase inhibited by citrate via competitive inhibition
-
Isomerization of citrate to isocitrate.
- Citrate isomerized to isocitrate by aconitase.
- ΔGº’ = 6.3 kJ/mol but reaction pushed to the right in vivo because product rapidly consumed in next step.
-
Oxidation of isocitrate to α-ketoglutarate and CO2.
-
Irreversible decarboxylation of isocitrate to α-ketoglutarate by isocitrate dehydrogenase.
- Inhibited by ATP and NADH.
- Stimulated by ADP and Ca2+
- Loss of CO2 make the reaction irreversible
- NADH produced
-
Irreversible decarboxylation of isocitrate to α-ketoglutarate by isocitrate dehydrogenase.
-
Oxidation of α-ketoglutarate to succinyl~CoA and CO2.
- Catalyzed by α-ketoglutarate dehydrogenase complex.
- Same family as PDH dehydrogenase.
- Cosubstrates (NAD and CoA)
- Coenzymes (TPP, FAD, and lipoic acid)
- Inhibited by succinyl~CoA and NADH
- Activated by Ca2+
- Not regulated by phosphorylation/dephosphorylation.
- NADH produced
- Loss of CO2 makes reaction irreversible.
- Catalyzed by α-ketoglutarate dehydrogenase complex.
-
Conversion of succinyl~CoA to succinate.
- Succinate thiokinase catalyzes the hydrolysis of the thioester bond in succinyl~CoA paired to the conversion of GDP to GTP.
- GTP interconvertible to ATP by nucleoside diphosphate kinase.
-
Oxidation of succinate to fumarate.
- Catalyzed by succinate dehydrogenase
- FADH2 made
- Enzyme located on inner mitochondrial membrane.
- ΔGº’ = 0 so reaction can go either way but pushed to the right in vivo because fumarate used in next step.
-
Regeneration of oxaloacetate.
- Fumarate converted to malate by fumarase.
-
Malate converted to oxaloacetate by malate dehydrogenase
- NADH made
- Rxn has a ΔGº’ = 29.7 kJ/mol but pulled to the right because OAA used by citrate synthase in step 1
- NADH made

Regulation of the TCA cycle
Regulated almost exclusively at the three irreversible steps.
- Citrate synthase competitively inhibited by citrate.
-
Isocitrate dehydrogenase
- Inhibited by ATP and NADH
- Stimuated by ADP and Ca++
-
α-ketoglutarate dehydrogenase
- Inhibited by succinyl~CoA and NADH
- Activated by Ca++
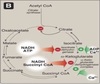
Additional roles of TCA cycle
TCA cycle intermediates can be used for other reactions such as:
- amino acid synthesis
- fatty acid synthesis
- gluconeogenesis
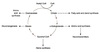
TCA cycle intermediate replenishment
Intermediates of the TCA cycle can be provided for by other metabolic pathways, specifically amino acid metabolism.
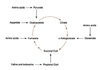
Electron transport chain
(ETC)
aka Respiratory chain
Basics
Inner mitochondrial membrane bound complex.
Consists of four seperate protein complexes.
Each complex accepts or donates electrons to/from mobile electron carriers (coenzyme Q and cytochrome C).
ETC pumps protons from matrix to intermembrane space to form a proton gradient.
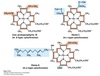
ETC complexes
-
Complex 1: NADH dehydrogenase
- Flavin mononucleotide (FMN) coenzyme accepts two electrons from NADH to become FMNH2.
- FMNH2 transfers electrons to CoQ to form CoQH2.
- Enzyme contains iron-sulfur (Fe-S) centers which acts as intermediate electron carriers.
-
Complex 2: Succinate dehydrogenase
- Electrons from succinate of TCA cycle transferred via Fe-S centers to FAD to form FADH2.
-
Complex 3: Cytochrome bc1 reductase
- Electrons from CoQH2 (from complex 1) are used to reduce cytochrome c which acts as electron carrier.
- Contains Fe-S centers which act as intermediates.
- Contains heme group which shifts from Fe3+ to Fe2+ and back as electrons move through.
-
Complex 4: Cytochrome oxidase
- Contains two different heme groups
- Heme a
- Heme a3
- Contains two Cu ions which act as intermediates
- Electrons from cytochrome c transferred to heme a via one of the Cu centers, then from heme a to heme a<em>3</em> via other Cu center, and finally to O2 to form H2O.
- Contains two different heme groups
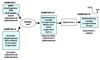
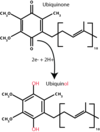
Coenzyme Q
Oxidized form ubiquinone reduced to ubiquinol after accepting electron.
Accepts electrons from complex I and II of the ETC.
Cytochromes
Contain a heme group in which the iron shifts from Fe3+ to Fe2+ oxidations states and back as electrons move to and from the heme group.
All contain the porphyrin ring with differing side chains.
Redox potentials
(E’º in volts)
Measures the ease with which an electron can be added or removed.
The more positive the value of E’º, the greater the tendancy of the oxidant in the redox pair to accept electrons.
Electrons flow from the redox pair with the more negative E’º to one with a less negative/more positive E’º.
Relationship of redox potential and free energy change
The redox potential (E’º) is related to the free energy change in the reaction by the Farady constant (F).
ΔG’º = -nFΔEº
n = number of electrons transferred
ΔEº = difference in reduction potentials of the overall reaction
Effective reduction potential
(E)
In the cell, the effective reduction potential (E) depends on the concentration of reactants.
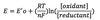
Electron flow in ETC
Due to the reduction potentials of each complex the electrons always flow downhill.
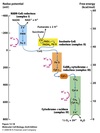
ETC site-specific inhibitors
Used to block electron flow in ETC.
Carriers befor the block become reduced and those after remain oxidized.

Oxidative phosphorylation
ATP synthase uses the proton gradient set up by ETC to produce ATP.
Dependent on the integrity of the inner mitochondrial membrane.
No high-energy intermediate exists (as with substrate-level phosphorylation).
Reactive oxygen species
(ROS)
When more electrons enter the ETC than can be immediately passed to O2 a state of oxidative stress exists.
Highly reactive superoxide free radicals such as superoxide (•O2-) are generated.
Mitochondria have systems to eliminate free radicals:
- Superoxide (•O2-) is converted to peroxide (H2O2) by superoxide dismutase.
- Peroxide converted to water by glutathione peroxidase.
NADPH used by glutathione reductase to regenerate glutathione peroxidase.
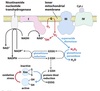
Respiratory control
The coupling of ATP synthesis and electron transport.
Neither process can occur without the other.
P:O ratio
The stoichiometry of ATP synthesis relative to the substrate that is oxidized.
The amount of ATP formed per ½ O2 consumed (or per pair of electrons.
Approaches 3 for NADH.
Closer to 2 for FADH2 (because succinate oxidation bypasses complex I)
Proton pumping by ETC
For each pair of electrons that travel through the ETC and transferred to O2:
Complex 1 pumps out 4 protons
Complex 3 pumps out 4 protons
Complex 4 pumps out 2 protons
Total of 10 protons per electron pair.
NADH + 11 H+M + ½ O2 → NAD+ + 10 H+IM + H2O
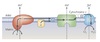
Proton motive force
(PMF)
Proton pumping by ETC across the inner mitochondrial membrane sets up a pH gradient (ΔpH) and a transmembrane potential (Δψ) with matrix side negative.
Can be calculated as
ΔG = 2.3 RT ΔpH + F Δψ
In general, transport of a pair of electrons through the ETC generates ~ 53 kcal.
Chemiosmotic Hypothesis
The proton motive force drives ATP synthesis as the protons flow passively back through the inner mitochondrial matrix down their concentration gradient through a proton pore in the ATP synthase.
The ~ 53 kcal produced per pair of electrons can drive the synthesis of 3 ATPs (using ~ 22 kcal) with the remaining energy used to drive ancillary reactions or dissipated as heat.
ADP + Pi + nH+IM → ATP + H2O + nH+M
Value of n depends on the structure of the ATP synthase and varies between species (~3.3 - 5 protons per ATP)
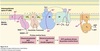
Mitochrondrial ATP synthase
(Complex V)
F1Fo ATPase
O stands for oligomycin which blocks the proton pore
F-type ATPase which functions as a reversible ATP-driven proton pump
- F1 portion contains the ATPase domain.
- Contains 9 subunits α3β3γδε
- α and β subunits for the knoblike catalytic section
- γ subunit forms a shaft that connects with Fo
- Fo complex consists of a, b, and c subunits
- 8 c-subunits form the c-ring in vertebrates
- Rotation of C-ring induces conformational changes in the β subunits of F1 that drive ATP synthesis
- Requires 8 protons moving across the membrane with each rotation and produces 3 ATP’s per turn
- 8 c-subunits form the c-ring in vertebrates

Yield of ATP production
- Takes 11 H+ to generate 3 ATP’s
- 8 H+ to give a complete c-ring rotation
- Additional 3 H+ to bring 3 phosphates into the matrix via the mitochondrial phosphate transporter
- From 1 NADH: 10 H+ pumped so (10/11) x 3 ATPs produced = 2.75 ATPs per NADH
- From 1 FADH2 (Succinate): 6 H+ pumped so (6/11) x 3 ATPs produced = 1.64 ATPs per FADH2
- Glycolysis generates 2 net ATPs and 2 NADH
- PDH generates 1 NADH/pyruvate → 2 NADH/glucose
- TCA generates:
- 3 NADH/pyruvate → 6 NADH/glucose
- 1 FADH2/pyruvate → 2 FADH2/glucose
- 1 ATP/pyruvate → 2 ATP/glucose
- OxPhos:
- 10 NADH → 27.5 ATP
- 2 FADH2 → 3.3 ATP
Going all the way through oxidative phosphorylation generates:
- ~ 31 ATP by OxPhos
- 2 ATP for glycolysis
- 2 ATP for TCA
TOTAL OF ~35 ATP/GLUCOSE
Respiratory inhibitors
CN- or CO
blocks electron transport and subsequently ATP synthesis
Oligomycin
Phosphorylation inhibitor
Blocks ATP synthesis and thus electron transport
Uncouplers
dinitrophenol (DNP)
uncoupling protein 1 (UCP1 aka thermogenin)
- Causes collapse of the proton motive force releasing energy in the form of heat rather than ATP.
- Electrons free to move through the ETC.
- Increases oxygen consumption.
Regulation of oxidative phosphorylation
ETC and ATP synthesis coupled so increasing or decreasing one does the same to the other.

Ancillary reactions
All the transport processes associated with oxidative phosphorylation which utilize the proton motive force or transmembrane potential.
Mostly used to transport molecules across the impermeable inner mitochondrial membrane.
- Agents that disrupt the PMF will reduce transport.
- Transport of these substances will conversely reduce the PMF available for ATP synthesis.
- Electroneutral transport systems driven by concentration gradients alone.
- Electrogenic transport systems utilize concentration gradients as well as the transmembrane potential.
- positively charged molecules enter matrix more easily
- Adenine nucleotide translocase
- Phosphate translocase
- Transporters for other charged metabolites such as pyruvate, malate, citrate, etc.
- Ca++ and asparate also transported in a PMF-dependent fashion
Adenine nucleotide translocase
Antiporter that exchanges ADP3- from the intermembrane space for ATP4- from the matrix.
Net transport of 1 negative charge out of the matrix (electrogenic) stimulated by the matrix-negative transmembrane potential.
Inhibited by atractyloside.
Phosphate translocase
Symporter that moves one phosphate (H2PO4- ) and one proton (H+) into the matrix.
Driven by the proton gradient established by ETC.
Electronically neutral.
ATP synthasome
Complex containing ATP synthase, adenine nucleotide translocase, and phosphate translocase can be isolated by gentle disruption of the inner mitochondrial membrane suggesting that these three proteins are spatially integrated.
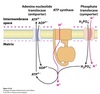
Nicotinamide nucleotide transhydrogenase
A transmembrane protein embedded in the inner mitochondrial membrane.
Uses the proton gradient to drive:
NADH + NADP+ + H+IM → NAD+ + NADPH + H+M
NADH is used by ETC so its levels are related to the potential for ROS generation.
Production of NADPH by transhydrogenase will increase as more electrons travel down ETC.
NADPH indirectly used in ROS neutralization.
Mitochondrial genes
Mitochondria contain their own genome which contains 37 genes.
13 encode subunits of respiratory chain proteins.
Mitochondrial diseases
Maternally inherited.
Defects in oxidative phosphorylation most commonly associated with mutations in mitochondrial genes.
Presumably due to generation of reactive oxygen species.
Affects tissues with high requirement for ATP such as brain, liver, and skeletal/cardiac muscle.
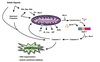
Mitochondria and apoptosis
- Triggered by:
- External signals acting via a receptor
- Oxidative stress
- Heat shock
- Viral infection
- Exposure to stimulus induces formation of large pores in the outer mitochondrial membrane called permeability transition complex
- Allows release of cytochrome c into the cytosol
- Cytochrome c in associated with apoptosis protease activating factor 1 (Apaf-1) activate a family of cytosolic proteases (the caspases) that degrade proteins and lead to cell death.
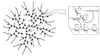
Glycogen
Structure & Function
- Major short-term storage form of carbohydrates in animals.
- For times of metabolic need.
- Branched chain homopolysaccharide of α-D-glucose
- Primary bond is α-1,4-glycosidic linkages.
- Every 8-10 glucose residues there is a branch attached via a α-1,6-glycosidic linkage
- Stored in the cytoplasm as large hydrated granules.
- Each granule contains as many as 55,000 glucose units.
- Found in the cytoplasm of liver and skeletal muscle cells primarily.
Sources of blood glucose
- Diet
- Degradation of glycogen
- Gluconeogenesis
Liver Glycogen
- Functions to maintain the blood glucose concentration particularly during the early stage of a fast.
- Glucose rapidly released from liver glycogen.
- Glucose able to enter systemic circulation due to presence of glucose-6-phosphatase in the liver.
- Hepatic glycogen stores ~24 hour supply of glucose.
- Liver also able to synthesize glucose via gluconeogenesis.

Skeletal Muscle Glycogen
- Serves as a fuel reserve for synthesis of ATP that will power muscle contraction.
- Muscle glycogen is not available to other tissues because muscle lacks glucose-6-phosphatase.

Methods of Activating Sugars
- Phosphorylation
- Glycolysis utilizes glucose-6-phosphate.
- Create a nucleotide sugar
- Glycogenesis utilizes UDP-glucose.
Alternate activation methods allows for both pathways to occur at the same time.
Glycogenesis
Step 1: Chain Synthesis
- Occurs in the cytoplasm and can be divided into two stages:
- Chain synthesis
- Chain branching
I. Chain Synthesis
A. Synthesis of UDP-glucose.
-
Glucose is phosphorylated to glucose-6-phosphate.
- By glucokinase in hepatic tissue.
- By hexokinase in peripheral tissue. - Glucose-6-phosphate converted to glucose-1-phosphate by phosphoglucomutase.
-
Glucose-1-phosphate reacts with UTP to form UDP-glucose and PPi which is catalyzed by glucose 1-phosphate uridylyltransferase (aka UDP-Glc pyrophosphorylase)
- Hydrolysis of PPi → 2 Pi by pyrophosphatase makes the reaction energetically favorable and irreversible.
B. Requirement of primer to initiate glycogen synthesis.
⇒Glycogen synthase cannot initiate glycogen synthesis de novo and can only add glucose to an existing chain, therefore, glycogenesis requires a primer.
- Glycogen fragment can serve as a primer for glycogen synthase which attaches glucosyl residues to existing chain using UDP-glucose.
- The protein glycogenin (homodimer) can prime glycogen synthesis and attach glucose residues through auto-glucosylation ⇒ serves as both substrate and enzyme in its role as primer.
a. The glycosyltransferase activity of glycogenin transfers the first molecules of glucose from UDP-glucose to a specific tyrosine side-chain (tyr-194) on itself.
b. After at least 4 (about 7) glucose residues have been added, glycogen synthase takes over.
- Glycogenin remains within the glycogen granule.
C. Elongation of glycogen chains.
- Glycogen synthase transfers glucose from UDP-glucose to the non-reducing end of the growing chain via α-1,4-linkages between the -OH group on C-1 of the activated sugar and the C-4 of the accepting sugar.
⇒ Glycogen synthase is the rate-limited and regulated enzyme of glycogenesis.
⇒ There are liver & muscle isozymes.
⇒ UDP released when α-1,4-glycosidic bond is formed can be converted to UTP by nucleoside diphosphate kinase + ATP.

Glycogenesis
Step 2: Chain Branching
- Occurs in the cytoplasm and can be divided into two stages:
- Chain synthesis
- Chain branching
II. Chain branching
A. Catalyzed by “branching enzyme” called glucosyl 4:6 transferase.
- Glucosyl 4:6 transferase cleaves an α-1,4-glycosidic bond from the non-reducing end of the glycogen chain producing a 6-8 glucosyl residues fragment.
- Enzyme then transfers the fragment to another residue on the linear chain via an α-1,6-glycosidic bond.
- Resulting new non-reducing end and old non-reducing ends can be further elongated by glycogen synthase.
- After further elongation, new chains of 6-8 residues can be transferred to make additional branches.
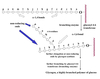
Functions of branching in Glycogen
- Increases solubility of glycogen molecule.
- Stored as hydrated granules.
- Increases number of non-reducing ends to which new glucosyl residues can be added to or removed from glycogen.
- Facilitates fast breakdown of glycogen into glucose when energy needed.
Glycogenolysis
Overview
- Occurs primarily in the cytoplasm of liver and skeletal muscle cells.
- Involves 2 stages:
- Shortening of chains
- Removal of branches
Glycogenolysis
Step 1: Shortening of Chains
-
Glycogen phosphorylase uses Pi to cleave the α-1,4-glycosidic bonds between glucose residues at the non-reducing ends of the glycogen chains releasing glucose-1-phosphate.
⇒Enzyme is a homodimeric exoglucosidase
⇒Requires pyridoxal phosphate (PLP) coenzyme (derivative of Vit B6)
⇒Liver and muscle isozymes- Glycogen phosphorylase stops attacking α-1,4-glycosidic bonds four glucosyl residues from an α-1,6-branch point.
- Resulting structure is called a limit dextrin.
- Glucose-1-phosphate is converted to glucose-6-phosphate by phosphoglucomutase.
- Next step in glycogenolysis depends on the tissue:
- In liver: glucose-6-phosphate is hydrolyzed to free glucose and Pi by glucose-6-phosphatase.
⇒Free glucose able to leave the liver and enter the blood stream. - In peripheral tissues: glucose-6-phosphate will be oxidized in the glycolytic pathway to produce energy.
- In liver: glucose-6-phosphate is hydrolyzed to free glucose and Pi by glucose-6-phosphatase.

Glycogenolysis
Step 2: Removal of branches
Catalyzed by a debranching enzyme which is a bifunctional protein with two catalytic activities.
-
4-α-D-glucantransferase activity transfers the outer three glucosyl residues of the limit dextrin to a non-reducing end by breaking and formation of α-1,4 bonds leaving one glucosyl residue in α-1,6 linkage.
⇒ 4,4 transferase activity - α-1,6 linkage is then cleaved hydroytically by the amylo-α-1,6 glucosidase activity of debranching enzyme releasing free glucose (non-phosphorylated).
- Co-operative and repetitive action of phosphorylase and debranching enzymes results in complete hydrolysis of glycogen to yield glucose-1-phosphate and free glucose in a 12:1 ratio.
⇒Free glucose is quickly phosphorylated in muscle for intracellular use.
⇒Phosphorylated glucose is already trapped inside muscle cells.
Regulation of Glycogen Metabolism
Key regulatory enzymes of glycogen metabolism:
Glycogen synthase (synthesis)
&
Glycogen phosphorylase (degradation)
Each is controlled by:
-
Hormone-induced covalent modification through phosphorylation or dephosphorylation of ser residues.
* Way of responding to the needs of the body as a whole. -
Allosteric effectors
* Way of responding to the need of a particular tissue at a particular time.
Glycogen Phosphorylase
Covalent Regulation
(Control of glycogen breakdown)
I. Regulation by covalent modification:
Glycogen phosphorylase exists in two forms:
A or active form: phosphorylated
B or inactive form: dephosphorylated
A and B forms are interconverted by:
Phosphorylase kinase
Produces active phosphorylated A-form.
&
Phosphoprotein phosphatase-1
Produces inactive dephosphorylated B-form.
*The A form of glycogen phosphorylase is more active because phosphorylation causes a conformational change in the enzyme which shifts the equilibrium of the enzyme between its T-state (taut and inactive) and R-state (relaxed and active) towards the active R-state. The B-form is inactive because the taut state is favored.
Phosphorylase kinase (regulatory enzyme) is itself regulated by phosphorylation/dephosphorylation.
A and B forms are interconverted by:
Protein Kinase A (PKA)
Produces active phosphorylated A-form.
&
Phosphoprotein phosphatase-1
Produces inactive dephosphorylated B-form.
Hormonal Control
of
Glycogen Breakdown
Hormonal signals that activate PKA include Glucagon and Epinephrine.
Phosphorylase kinase (regulatory enzyme)
&
Glycogen phosphorylase (regulated enzyme of glycogenolysis)
are phosphorylated in response to hormonal signals that are transduced via cAMP which activates protein kinase A.
PKA also results in the inhibition of phosphoprotein phosphatase-1 by phosphorylating and activating protein phosphatase inhibitor (A-form).
This enzyme is used to maintain the level of phosphorylation until hormone signal changes.
Active protein kinase A has a short half-life because seperation of regulatory and catalytic subunits revealed PEST sequences which marks protein for degradation.

Glucagon
&
Glycogen Regulation
Peptide hormone released from the alpha-cells of the pancreas when blood glucose is low.
Glucagon binds to plasma membrane receptors on liver cells but not muscle.
Stimulates glycogen degradation via PKA-mediated activation of phosphorylase kinase.
Activated during periods of fasting thus making glucose available to tissues.
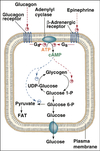
Glycogen Synthase
Covalent Regulation
(Control of glycogen synthesis)
Glycogen synthase exists in two forms:
A or active form: dephosphorylated
B or inactive form: phosphorylated
Several kinases phosphorylate glycogen synthase A to the inactive B form including:
- cAMP dependent PKA*
- Phosphorylase kinase*
- Glycogen synthase kinase-3*
- AMP-dependent kinase*
B-form converted back into the active A-form by
Phosphoprotein phosphatase-1.
PKA also results in the inhibition of phosphoprotein phosphatase-1 by phosphorylating and activating protein phosphatase inhibitor (A-form).
Reciprocal Regulation of Glycogen
Synthesis & Degradation
cAMP regulates glycogen metabolism through the simultaneous activation of glycogenolysis and inhibition of glycogenesis.
Also displays amplification: 1 hormone activates many adenyl cyclase, makes many cAMP, each activates many PKA, each phosphorylates many phosphorylase kinase, each phosphorylates many phosphorylase ect.

Insulin
Released by pancreas in response to high blood glucose levels.
Has the opposite affect of glucagon/epinephrine.
Activates glycogen synthesis and inhibits degradation in liver and muscle.
- Promotes inhibition of several protein kinases and activation of phosphoprotein phosphatase.
- Causes subsequent dephosphorylation of glycogen synthase (activating) and dephosphorylation of phosphorylase kinase and phosphorylase (inactivating).
- Promotes conersion of cAMP to 5’ AMP by activating a phosphodiesterase.
- Causes subsequent decrease in active PKA.
Allosteric Regulation of Glycogen Synthesis
- Allosteric effectors are superimposed onto covalent regulation in order to meet the needs of the tissue.
- Enzymes of glycogen metabolism including glycogen phosphorylase kinase, glycogen phosphorylase, and glycogen synthase are regulated in an allosteric effectors acting in a non-covalent manner.
- Postive allosteric effectors bind to a regulatory site on the R (active) form of the enzyme stabilizing it and pulling the equilibrium towards the R-form.
- Negative allosteric effectors bind to the T (inactive) form and stabilizes that pulling equilibrium towards T-form.
- Effectors include:
- Ca2+ and AMP which are signs of low energy
- Glucose and glucose-6-phosphate which are signs of high energy
Calcium Mediated
Regulation of Glycogen Metabolism
Released in times of energy need.
Ca2+ binds to the calmodulin-like δ-subunits of dephosphorylated (B-form) phosphorylase kinase causing conformational change which activates the catalytic γ-subunits in the absence of phosphorylation ⇒ stabilizes the R-state
Ca2+ also required for maximal activation of phosphorylase kinase a.
Phosphorylase kinase subsequently phosphorylates and inhibits glycogen synthase.
End result of a rise in [Ca2+]in is increased degradation and decreased synthesis of glycogen.
-
Contracting muscle
- Ca2+ released in in response to nerve impulses.
- [Ca2+]in activates sarcoplasmic glycogenolysis by activating phosphorylase kinase.
-
Liver cells
- Epinephrine binds to α-adrenergic receptors, activating phospholipase-C, and generating IP3 and DAG from PIP2.
- IP3 causes release of Ca2+ from the SER.
- [Ca2+]in activates glycogenolysis by activating phosphorylase kinase⇒ activates glycogen degradation.
- DAG activates PKC which phosphorylates and inactivates glycogen synthase ⇒ inhibites glycogen synthesis.
- IP3 causes release of Ca2+ from the SER.
- Epinephrine binds to α-adrenergic receptors, activating phospholipase-C, and generating IP3 and DAG from PIP2.
Side note: Ca2+ also activates mitochondrial events:
- Activates PDH phosphatase thereby activating PDH and pyruvate degradation.
- Allosteric effector of isocitrate dehydrogenase and α-ketoglutarate dehydrogenase thereby activating TCA cycle.
AMP Mediated
Regulation of Glycogen Metabolism
- AMP to ATP ratio reflects the energy state in muscle cells.
- Increase in AMP signals low energy and need for glycogen degradation.
- AMP functions as an allosteric activator of muscle phosphorylase b.
- Directly activates the dephosphorylated form of myophosphorylase b.
Glucose Mediated
Regulation of Glycogen Metabolism
- When glucose is plentiful, heptatic glycogenolysis is decreased by glucose itself acting as an allosteric inhibitor of hepatic phosphorylase a.
- The glucose-6-phosphate fromed from glucose is an allosteric activator of glycogen synthase b in both liver and muscle, thus increasing glycogenesis.
- For this reason, glycogen synthase b is sometimes designated “D” because it is dependent on glucose-6-phosphate for activity while synthase a is designed “I” for independent.
GSD Type Ia
Von Gierke Disease
-
Glucose-6-phosphatase deficiency
- Liver and kidney
- Severe fasting hypoglycemia hallmark
- Major Findings
- Hepato/renomegaly
- Fasting hypoglycemia
- Lactic acidemia
- Hyperuricemia
- Hyperlipidemia
GSD Type II
Pompe Disease
aka
Generalized Glycogenosis
- Lysosomal acid α-glucosidase deficiency
- Infantile, juvenile, and adult-onset forms
- Affects all organs but skeletal/cardiac most
- Cardiomegaly/myopathy in infantile forms
- Muscle weakness in later forms
- Enzyme replacement therapy has reduced mortality
GSD Type III
Cori Disease
aka
Limit Dextrinosis
- Debranching enzyme deficiency (both actions)
- IIIa : affects liver and muscle
- IIIb: affects liver only
- Milder hepatomegaly
- Muscle weakness
- Accumulated glycogen has abnormal structure with shorter chains
- May cause liver fibrosis or cirrhosis
GSD Type IV
Andersen Disease
aka
Amylopectinosis
- Branching enzyme deficiency in liver
- Progressive hepatomegaly
- Accumulated glycogen has abnormal structure with longer chains and no branches
- Progressive liver cirrhosis in infantile form can be lethal
GSD Type V
McArdle Disease
- Muscle glycogen phosphorylase deficiency
- Infantile and Adult forms
- Exercise intolerance and muscle cramps
- Symptoms usually first appear in adolescence
- Failure of blood lactate to rise after anaerobic exercise
GSD Type VI
Hers disease
- Liver glycogen phosphorylase deficiency
- Hepatomegaly
- Benign in general
- Mild fasting hypoglycemia
GSD Type VII
Tarui Disease
- Muscle PFK-1 deficiency
- Affects muscle and RBC’s
- More severe exercise intolerance and muscle cramps
- RBC’s show some percentage of normal activity
Gluconeogenesis
Basics
- The synthesis of glucose from smaller precursors such as:
- pyruvate
- lactate
- glycerol
- most amino acids
- Alanine ⇒ pyruvate
- Glutamine ⇒ α-ketoglutarate
- Except: leucine and lysine
- Serves to stabilize glucose levels in the blood during fasting after glycogen stores have been depleted
- Occurs in the liver and to some extent the kidneys
- Requires specialized gluconeogenic enzymes to bypass the irreversible steps of glycolysis

Gluconeogenesis
Bypass 1
-
Pyruvate + HCO3- → oxaloacetate
by pyruvate carboxylase + ATP
Reaction is anaplerotic because it yields a citric acid cycle intermediate OAA.
2. Oxaloacetate → PEP + CO2
by phosphoenolpyruvate carboxykinase (PEPCK) + GTP
Required to bypass the highly exergonic reaction catalyzed by pyruvate kinase during glycolysis.

Pyruvate Carboxylase
Mechanism
- Biotin-containing multifunctional protein
- Biotin coenzyme linked to a lysine residue
During course of reaction:
- Carbon dioxide is first bound to the biotin in one active site forming a high energy carboxy-biotin species.
- Long flexible side chain swings the carboxy-biotin to the second active site.
- Carboxy-group transferred to pyruvate.
Pyruvate Carboxylase
Regulation
Allosterically activated by acetyl CoA.
Acetyl CoA is a product of fatty acid metabolism.
Links gluconeogenesis and fat catabolism.
Acetyl CoA also inhibits pyruvate dehydrogenase by activating PDH kinase.
Decreased glycolysis, increased gluconeogensis, and increased fatty acid catabolism by the liver when blood glucose low.
Compartmentalized in the mitochrondria.
Gluconeogenesis
Bypass 2
Fructose-1,6-bisphosphate → fructose-6-phosphate
by fructose-1,6-bisphosphatase
By passes the reaction of PFK1 in glycolysis.
fructose-1,6-bisphosphatase
Allosterically inhibited by F-2,6-P2 and AMP
Same allosteric activators of PFK1 = Reciprocal regulation
Gluconeogensis
Bypass 3
Glucose-6-phosphate → glucose
by glucose-6-phosphatase
By passes reaction of hexokinase/glucokinase.
Allows glucose to leave the cell and enter the blood.
Glucose-6-phosphatase
Glucose-6-Phosphatase is a membrane bound enzyme located in the ER.
Active site faces into the lumen of ER.
Glucose-6-P must enter then glucose and Pi must leave.
Deficiency causes glycogen storage disease Ia.
Found only in liver and kidney.
There is no direct control of the enzyme but has a high KM for glucose-6-P so only functions when concentrations high.
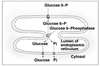
Reciprocal Regulation
of
Glycolysis and Gluconeogensis
Glycolysis converts 1 glucose into 2 molecules of pyruvate.
Produces 2 NADH and 2 ATP
Gluconeogenesis converts 2 pyruvates into 1 glucose
Consumes 2 NADH and 6 ATP or GTP
Two processes are tightly regulated so that only once may proceed at a time.
Allosteric Regulation
Important sites for control are the irreversible reactions
- pyruvate kinase - F-1,6-P2 (+) , ATP (-)
pyruvate carboxylase - Acetyl CoA (+), ADP (-) - phosphofructokinase - AMP (+) , F-2,6-P2 (-)
fructose bisphosphatase - AMP (-) , F-2,6-P2 (-)
Hormonal Regulation
- Glucagon → increased cAMP → phosphorylation of hepatic pyruvate kinase and PFK 2 → decrease glycolysis
- Insulin → decreased cAMP → increase hepatic glycolysis
Transcriptional Regulation
- Glucagon → stimulates expression of PEPCK and maybe glucose-6-phosphatase through phosphorylation of cAMP response element (CREB) by PKA
- Insulin → stimulates expression of PFK1, pyruvate kinase, PFK2, enzymes of glycolysis

Compartmentalization of Gluconeogenesis
Gluconeogenesis requires both mitochrondrial and cytosolic enzymes.
Pyruvate carboxylase is a mitochondrial enzyme while the other gluconeogenic enzymes are largely cytoplasmic.
Pyruvate must be transported into the mitochondria where it is converted to OAA.
OAAmito ⇒ Malatemito ⇒ Malatecyto ⇒ OAAcyto
Gluconeogenesis consumes NADH in the cytosol in the conversion of 1,3-bisphosphoglycerate to glyceraldehyde-3-P but the NADH/NAD+ ratio is normally very low so glycolysis usually preferred.

Gluconeogenesis from lactate
- Occurs in the liver
-
Lactate converted to pyruvate in the cytosol by LDH
- Yields NADH ⇒ export of reducing equivalents fro mthe mitochondria is not nescessary
- Pyruvate enters mitochondria where it is converted to OAA by pyruvate carboxylase
- OAA can either:
- Be converted to aspartate by transamination in mitochondria ⇒ leave mitochondria via aspartate shuttle ⇒ converted back to OAA in the cytosol
- Be converted to PEP by mitochrondrial PEP carboxykinase ⇒ PEP leaves mitochondria

The Cori Cycle
Recycles glucose carbons from lactate in order to maintain blood glucose levels.
Lacate produced via anaerobic glycolysis travels to the liver.
There it is used for gluconeogenesis and resulting glucose is released back in to the blood.
(Liver hopes the next time the glucose will be used oxidatively for more energy.)
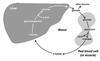
Base?
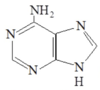
Adenine
Base?

Guanine
Base?

Xanthine
Base?
Hypoxanthine
Base?
Thymine
Base?

Uracil
Base?
Orotic Acid
Purine De Novo Synthesis
Overview
Built on a ribose-5-phosphate.
Occurs primarily in cytosol of hepatocytes.

Purine Synthesis
C & N
Sources and Addition Order
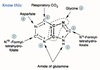
Formation of IMP
Mechanism
11 step process
Requires a large amount of ATP
-
Ribose-5-P ⇒ PRPP by PRPP synthetase
- Regulated but not committed step
- Add N from glutamine to PRPP by PRPP glutamyl amidotransferase
- Committed step
- Highly regulated
- Rate-limiting
- Add glycine backbone
- Add C from N10-THF
- Add N from glycine for ring 2
- Ring 1 closure
- Add C from respiratory CO2
- Attach aspartate R group N to C of respiratory CO2
- Keep only N from aspartate and release backbone as fumarate
- Add C from N10-THF
- Close ring 2 to form inosine monophosphate (IMP)
PRPP Synthetase
Regulation
Catalyzes the regulated but not committed step of purine de novo synthesis.
Feed back inhibition by purine ribonucleotides (mainly AMP and GMP)
Activated by Pi
PRPP Glutamyl Amidotransferase
Regulation
Catalyzed the committed and rate-limiting step of purine de novo synthesis.
Inhibited by end-products (purine ribonucleotides, inactive dimer)
Activated by PRPP and active monomer
High [PRPP] able to overcome end-product inhibition.
IMP ⇒ AMP & GMP
IMP converted to AMP or GMP in seperate two step reactions.
IMP ⇒ AMP
IMP + Asp ⇒ Adenylosuccinate monophosphate (ASMP)
ASMP ⇒ AMP + Fumarate
IMP ⇒ GMP
IMP ⇒ XMP
XMP + Gln ⇒ GMP + Glutamate
IMP ⇒ AMP or GMP
Regulation
Reciprocal regulation ensures appropriate levels of AMP and GMP.
1st step in IMP ⇒ AMP inhibited by AMP and requires GTP.
2nd step in conversion of IMP ⇒ GMP inhibited by GMP and requires ATP.

Adenine and Guanine
Interconversions
Meets the needs of the cell.

Ribonucleotide Reductase
Maintains adequate and balanced concentrations of deoxyribonucleotides.
Catalyzed by ribonucleotide reductase.
Requires reduced thioredoxin coenzyme.
RNR has 3 sites:
- Active site ⇒ reduces ribonucleotide to deoxyribonucleotide
-
Activity site ⇒ on/off switch
- ATP activates
- dATP inactivates
-
Substrate specificity site ⇒ binds a specific positive allosteric effector (NTP or dNTP) to control reductive of a given NDP
- C ⇒ U ⇒ G ⇒ A
- Ex. Binding of dCTP makes enzyme specific for UDP
RNR inhibited pharmacologically by hydroxyurea.

Nucleotide Catabolism
Big Steps

Purine Catabolism

Uric Acid Production
Mechanism
Guanine ⇒ Xanthine by Guanase
Adenosine ⇒ Inosine by Adenosine Deaminase
Inosine ⇒ Hypoxanthine by Purine nucleoside phosphorylase
Hypoxanthine ⇒ Xanthine by Xanthine Oxidase
Xanthine ⇒ Uric acid by Xanthine Oxidase

Purine
Salvage Pathways
Free purine bases or nucleosides can be reutilized.
Important in non-hepatic tissues.
Base salvage:
Adenine phosphoribosyltransferase (APRT)
Adenine + PRPP ⇒ AMP + PPI
Hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
H or G + PRPP ⇒ IMP or GMP + PPi
Nucleoside salvage:
Adenosine kinase
Adenosine + Pi ⇒ AMP
Salvage Effects
on
De Novo Purine Synthesis
Salvage inhibits de novo synthesis:
Decreases [PRPP] ⇒ base salvage only
Generates nucleoside monophosphates that inhibit the amidotransferase ⇒ base and nucleoside salvage
ADA Deficiency
Adenosine Deaminase (ADA) Deficiency
⊗ ADA ⇒ ⊗ adenosine catabolism ⇒ ↑ [dATP] ⇒ ⊗ RNR ⇒ developmental arrest and/or apoptosis of lymphocytes
Results in severe combined immunodeficiency syndrome ⇒ ADA-SCIDS
PNP Deficiency
Purine nucleoside phosphorylase deficiency
Rarer and less severe than ADA deficiency.
Primarily affects T-cells.
Lesch-Nyhan Syndrome
- X-linked
- Hypoxanthine-Guanine Phosphoribosyltransferase (HGPRT) deficiency
- ↑ [PRPP] ⇒ ↑ [uric acid]
- Symptoms:
- hyeruricemia with gout
- compulsive self-mutilation
- aggressive behavior
- developmental delay
- profound motor impairment
- early death from renal failure
Pyrimidine De Novo Synthesis
Atom Sources
Build the base directly then attach to a sugar phosphate.
Pyrimidine De Novo Synthesis
Overview

Pyrimidine De Novo Synthesis
To UMP Level
Mechanism
Pyrimidine ring made then linked to ribose phosphate.
CAD catalyzed first 3 reactions.
UMP synthase catalyzes reactions 5 & 6.
-
Orotic acid is the first pyrimidine formed
- Regulated and rate-limiting step is synthesis of carbamoyl phosphate by CPS II.
- Committed step is synthesis of carbamoyl aspartate by aspartate transcarbamoylase.
- Orotic acid + PRPP ⇒ OMP (first nucleotide)
- OMP decarboxylated to UMP

UTP ⇒ CTP
Mechanism
OMP ⇒ UMP
- UMP phosphorylated by kinases to UDP and UTP
- UTP aminated using Gln to CTP and Glu by CTP synthetase

CTP Synthetase
Regulation
⊕ UTP
⊖ CTP
dUMP synthesis
Mechanism
UDP ⇒ dUDP by RNR
dUMP arises from both dUD(T)P and dCMP
dUDP ⇒ dUTP by kinase
dUTP ⇒ dUDP ⇒ dUMP by dUTPase
dCMP ⇒ dUMP by deamination

dUMP ⇒ TMP
Mechanism
dUMP methylated at the 5 position by thymidylate synthase ⇒ TMP
Thymidylate synthase inhibited by 5FU.
Uses N5N10-methylene THF as 1 C donor.
THF oxidized to DHF as methylene group reduced to methyl group.
DHF reduced back to THF by dihydrofolate reductase + NADPH.
Dihydroflocate reductase inhibited by methotrexate.
Traps folate as DHF ⇒ ↓ thymidylate synthesis ⇒ ↓ DNA synthesis
5-FU
5-FU converted to nucleotide monophosphate by addition of PRPP.
Deoxy from of nucleotide inhibits thymidylate synthase.

Methotrexate
Folic acid analog
Inhibits dihydrofolate reductase.
↓ thymidylate synthesis ⇒ ↓ DNA synthesis
Used to treat some cancers and psoriasis.
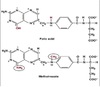
CAD Protein
Trifunctional protein.
Catalyzes the first 3 steps of pyrimidine synthesis:
- Carbamoyl phosphate synthetase II
- Aspartate transcarbamoylase
- Dihyrorotase
Pyrimidine Synthesis
Regulation
Carbamoyl phosphate synthetase II is the regulated enzyme
⊕ PRPP
⊖ UTP
Hereditary Orotic Aciduria
UMP synthase deficient
- Orotate phosphoribosyl transferase
- Orotate decarboxylase
↑ [orotic acid] and ↑ excretion of OA
Symptoms are failure to grow and develop and anemia.
Treat with uridine:
Uridine → UMP →→ UTP → CTP
Requires functional salvage pathway.
UTP inhibits CPSII of pyrimidine de novo synthesis so decreases production of OA also.
Pyrimidine
Catabolism and Salvage
All products are water soluble.
There are no known pathologies of salvage or degradation.
Nucleotide → Nucleoside → Free base:
Cytosine deaminated to uracil.
Uracil → β-alamine, CO2 and NH4
Thymine → β-aminoisobutyrate, CO2 and NH+
Some salvage of nucleosides and bases to nucleotides.
β-aminoisobutyrate
Thymine → β-aminoisobutyrate, CO2 and NH+
β-aminoisobutyrate is unique to thymine degradation and urinary excretion used as a measure of DNA turnover
Gout
Definition & Description
Disorder of purine metabolism.
Requires:
Hyperuricemia
Precipitation of uric acid or monosodium urate crystals in tissues
Inflammatory response
Tophi may be seen.
Monosodium Urate
Uric acid deprotinated at physiological pH to urate
Urate + sodium ions ⇒ monosodium urate
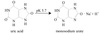
Gout
Pathophysiology
-
Hyperuricemia
- Necessary precondition for gout but not enough
- All patients with gout show hyperuricemia but only minority of people with hyperuricemia experience gout
-
Formation and deposition of urate crystals
- Tendency for crystal deposition varies
- Factors may precipitate crystal formation
- ↓ temperature
- Explain why gout most commonly affects great toe (podagra)
- ∆ pH
- trauma
- stress
- ↓ temperature
- Inflammatory response to the crystals
Clinical Stages
of
Gout
Asymptomatic hyperuricemia
↓
Acute intermittent attacks
↓
Chronic tophaccous gout
May see urolithiasis.
Primary Gout
Gout is the major manisfestation of an innate disorder.
>90% of cases hyperuricemia due to ↓ excretion not ↑ production
Most cases idiopathic.
Known molecular defects:
- PRPP synthetase with increased activity ⇒ super synthetase
- Partial deficiency of HGPRT
Secondary Gout
Gout is a secondary manifestation of an underlying disorder.
- Total deficiency (<1.5% of normal) of HGPRT seen in X-linked Lesch-Nyhan syndrome.
- Increased cell turnover
- cancers
- hemolytic diseases
- psoriasis
- chemotherapy
- Decreased renal excretion
- lead poisoning
- Increased conversion of ATP ⇒ AMP
- ETOH abuse
- Type I glycogen storage disease
- Inborn errors of fructose metabolism
- Hypoxia
Treatment of Acute Gout
Therapy to reduce inflammation.
No effect on urate concentrations.
-
NSAIDS
- Indomethacin
- Aspirin contraindicated
- competes with urate for renal excretion
-
Colchicine
- Inhibits phagocytosis of urate crystals
- Inhibits microtubule assembly
- Many side effects
- Low dose used prophylactically
- Inhibits phagocytosis of urate crystals
-
Steroids
- Glucocorticoids
Treatment of Chronic Gout
Life-long therapy to decrease urate concentration.
No direct effect on inflammation.
- Increasing renal excretion ⇒ uricosuric agents
-
Probenecid
- Inhibits renal reabsorption of urate
-
Probenecid
- Decreasing urate synthesis
-
Xanthine oxidase inhibitors
- Allopurinol
- Febuxostat
-
Xanthine oxidase inhibitors
-
Uricase
- urate to more soluble allantoin
