Week 11.19: DNA to Physiology Flashcards
DNA to Physiology
Learning Objectives
- Explain how links are found between DNA variation and physiological levels of biochemical entities (e.g proteins, metabolites)
- Define what is meant by expression quantitative trait loci (eQTL)
- Describe how databases of eQTLs and can be used to infer the effect of novel and rare variants on an individual’s physiology
- Identify the limitations of current methods for relating genomic variation to physiology and propose how these may be overcome in the future
Introduction
**Haven’t we been here before? **
We have looked at genome-wide association studies (GWAS), which can reveal relationships between genomic variants and traits of interest (e.g disease risk).
But GWAS has two important limitations;
1. By definition, such studies only reveal associations – they tell us nothing about the underlying physiology
2. GWAS can only provide statistically significant results for alleles which are carried by a sufficient number of people within the study population
1.By definition, such studies only reveal associations – they tell us nothing about the underlying physiology
For example:
Presence of specific alleles increased risk of diabetes
What is happening here within the body’s biological pathways, organs, etc.?

2. GWAS can only provide statistically significant results for alleles which are carried by a sufficient number of people within the study population
“”Rare” variants (especially rare combinations of variants) are actually very common, so this is important. Previously unseen “novel” variants can occur whenever a new individual is conceived.
Physiology and genomic variations
One way to better understand how genomic variation gives rise to phenotypic traits is via this two-step process?
<!--[if !supportLists]-->
1> ** <!--[endif]-->Determine how genetic variations affect the abundance of key biomolecules, e.g** transcripts, proteins, metabolites
<!--[if !supportLists]-->
2> ** <!--[endif]-->Study biological pathways to see how changes to the abundance of the affected molecules can affect the phenotype.**
1>Study biological pathways to see how changes to the abundance of the affected molecules can affect the phenotype.
Key to this process is the concept of expression quantitative trait loci (eQTLs)
1.1A little background: GTLs
Quantitative traits
Quantitative traits
The GWAS examples we’ve looked at so far have mostly been aimed at finding associations between genetic variants and the presence or absence of particular phenotypic traits (e.g disease susceptibility) These are known as discrete traits.
Some traits are inherently quantitative or continuous, e.g height and other morphological characteristics.
Continuous traits are often due to multiple DNA variants. These are often different genes (i.e they are polygenic)

Continuous traits are often due to multiple DNA variants. These are often different genes (i.e they are polygenic)
Why?
A simple analogy:
A single light switch can only give you two states: on/off
Combining multiple switches give you a range of different light levels

QTLs (Quantitative Traits Loci)
Chromosomal regions that underlie continuous traits are known as quantitative traits loci (QTLs)
QTLs are much studied due to many important commercial applications e.g breeding plants and animals for maximum yield, etc.
Agricultural researchers have the freedom to breed organisms with specific traits.
In human studies we have to build experiments from the available population, example;
[IMAGE]
There’s a clear quantitative relationship between genotype (at marker locus rs6749447 in this case) and phenotype (systolic blood pressure)
To infer a relationship for a given locus-trait pair, we need to assess the statistical significance of the difference between the mean trait values for the two genotypes. This is an example of a quantitative trait – simple example

Application of a T-test or similar is a good solution here
Statistical tests used
If a DNA marker is not linked to a QTL, then the mean values of the phenotypic trait will not vary among individuals with different genotypes at the maker locus

Finding QTLs – Osteoporosis as a case study
Osteoporosis is characterised by low bone mineral density (a quantitative trait)
A 2000-wide study sought to link genetic loci with BMD in a human population
PubMed: 10999795
**How did they do it? **
- . <!--[endif]-->First of all, assemble a good sized population (595 US citizens pairs comprising 464 Caucasians and 131 African-Americans, with detailed medical history (e.g fracture, therapy information) and genotyping data
- <!--[if !supportLists]-->
- <!--[endif]-->Collect accurate bone density measurements for these people, done using DEXA (an X-ray technique).
- Correct BMD values for age and gender, because it varies massively according to these factors
- Carry out QTL mapping, using genome wide linkage analysis (lectures 7 & 8)
In simple terms, this means seeking statistically significant relationships (for this study LOD score . 1.85) between genetic features and measured BMD.
QTLs alone cannot accurately predict quantitative traits, because environmental factors (GxE effects) also contribute to the trait. For example, BMD is affected by calcium intake
Great introduction to QTL mapping and its applications: PubMed: 19584810
eQTLs
To move from phenotype to physiology, we need to consider expression quantitative trait loci (eQTLs)
An eQTL is a statistical association between a genomic locus and the expression level of a particular gene transcript. The protein equivalent is called a pQTL and for metabolites mQTL
Discovering eQTLs is very similar to discovering QTls for phenotypic traits, expect that we need expression data for each person. We already saw methods for collecting this data (lectures 9 & 10).

Human skin case study
A 2010 study identified 841 cis-acting eQTLs through the analysis of skin from 110 people – 53-psoriatic, 57 healthy controls PubMed ID: 211297226
Genotype data was acquired using SNP arrays,
Biopsies were taken from lesional and non-involved skin and gene expression data acquired using ~54,000 probe microarrays
For each gene, associations were sought between SNPs within 1Mb of the gene transcription start site and 1Mb of the transcription end site
This kind of targeting means we can get statistically significant results in much smaller populations than if we were to look genome-wide
Regional plots for evidence of cis-association between SNPs & ERAP2 or RPS26
The most significant SNPs are highlighted with a square. The other SNPs are drawn as circles and colour coded according to the degree of linkage disequilibrium (i.e likely association) with the most significant SNP.
The most significant SNPs are highlighted with a square. The other SNPs are drawn as circles and colour coded according to the degree of linkage disequilibrium (i.e likely association) with the most significant SNP.

Extracting the most significant SNPs looks like:

How can we use eQTLs?
Given the genome of an individual, we can map known eQTLs to it, this will tell us which gene transcripts are likely to be affected by the individual’s particular genotype
Using eQTLs and to infer physiological variation
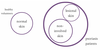
Of most interest are those alleles that are relatively uncommon (e.g <5% population frequency) since there effects are unlikely to be known already, we can find this frequency information from HapMap or similar variation projects.
It’s this subset of alleles that we can most usefully work with
For each allele, we can look at the gene whose transcription of the gene whose transcription is affected by that allele, and knowing the function of that gene (from some database) we can deduce phenotypic traits that may result.
For example, if an eQTL is known to reduce the transcription of CYP2E1, that will lead to a deficit of CYP2E1 protein, reducing the body’s ability to metabolise ethanol and various pharmaceuticals.
eQTL enrichment analysis
Of more interest are the more difficult to detect emergent effects caused by multiple genetic variations
A particular combination of variants may be so rare it may never be seen in a population with a frequency high enough to find a statistically significant association in a GWAS
Finding these kinds of relationships is down by enrichment analysis
Enrichment analysis is essentially process for comparing gene lists to see if a set of genes play a significant role in a particular process or pathway
KEGG – genome pathways
What is GO?
GO (gene ontology) is a effectively a community agreed dictionary of terms for describing:
- Biological process
- Cellular component
- Molecular function
Example term: insulin secretion
Gene products with kwon function are annotated with these terms, e.g Uniprot
But its more than that …
GO terms are organised hierarchically so if we select genes according to a higher level term, child terms can also be included.

What is Reactome?
Reactome is a database of manually curated peer reviewed biological pathways
For example, the insulin receptor signalling cascade:
[IMAGE]
You can interact with the pathway on screen like this, or (as in the case of gene enrichment) access the information programmatically.

Once you have your gene lists, there are numerous bioinformatics that can be used for the enrichment, e.g WebGestalt

11.19 Going deeper
Beyond associations… to physiological insights
Gene enrichment based eQTLs has its limitations:
There are not very many well characterised eQTLs available in the public domain
The enrichment only shows us associations with certain pathways or functions – it doesn’t reveal the effects of variants on underlying biological processes
As research scientists (rather than consumer or clinicians) we may want to dig deeper into the associated pathways in KEGG or Reactome to see exactly what effect a particular genotype has.
RegulomeDB
We might also want to look at the specific relationship between SNPs and regulatory elements (e.g transcription factor binding sites, promoter regions).
RegulomeDB is one source of information for this – with associations found by ENCODE and other projects
Via the web interface, you can search for information about at given variant an a score is given according to level of support for this having a regulatory effect
Most of the TF binding evidence comes frm ChIP-seq (Chromatin ImmunoPrecipitation Sequencing) experiments
The eQTL information is from transcriptomics experiments –
A RegulomeDB search can be a good starting point in gaining a detailed understanding of how a SNP affects physiology


