Overview and Embryology Flashcards
Efferent neurone columns
3
Somatic
Branchial
Visceral
Arrangement of efferent cell columns from M->L
SBV
Somatic
Branchial
Visceral
Afferent neurone columns
Special visceral
General visceral
Special somatic
General somatic
Arrangement of afferent cell columns from M-> L
VG VS, SG, SS
Visceral General
Visceral special (taste)
Somatic general
Somatic special (hearing, vestibular)
To what do the colours correspond
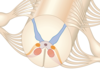
Blue- sensory
Orange- intermediant visceral (i.e. autonomic)
Red motor
Pure corticospinal tracts in humans
Thought to be exceedingly rare due to close proximity to other corticofugal tracts (corticoreticular, corticopontine, reticulospinal, vestibulospinal).
Thought to result in deficits in delicate fractionate movement and Babinski
Fucntion of ventral rami of spinal nerves
Innervates limbs and anterior skin and muscles of trunk
Function of dorsal rami of spinal nerves
Postvertebral muscles and skin of back
nAChR
Inotropic receptor
mAChR
Metabotropic receptor (GPCR)
Formation of the cerebelleum
Forms from dorsolateral thickenings of metencephalon which overgrow the root of the fourth ventricle (rhombic lips)
These lips fuse in the midline to form the cerebellar vermis.
Peripheral neuroblasts contribute to cerebellar cortex, central-> deep nuclei

Origin of tectal nuclei
Neuroblasts from the alar plates migrate into the tectum to form the superior and inferior colliculi
Origin of central gray matter around aqueduct
Neurobalsts of the alar plates
What are the three major commissures beign to develop in the lamina terminalis
Anterior commissure
Hippocampal commissure
Corpus callosum
Describe the development of the corpus callosum
7th week
The dorsal aspect of the lamina terminalis thickens into the commissural plate which becomes thickened with cellular material that forms a glial bridge across the groove
Develops in rostral to caudal fashion.
The exception is the rostral most portion of the corpus callosum- rostrum and anterior genu which develop last

What might cause violation of the corpus callosal front to back development
Secondary destructive processes might damage the corpus callosum after it is already formed
How does the presentation of corpus callosum agenesis occur?
Due to front to back development, then developmental arrest will normally result in an intact genu with a partially or completely formed body and small or absent splenium or rostrum.
Small or absent genu but intact splenium and rostrum in corpus callosal agenesis suggests?
Secondary destructive process
What is the corpus callosum abnormality which is an exception to the front to back and secondary destructive lesion
The callosal abnormality associated with holoprosencephaly in which the corpus callosum demonstrates and intact splenium in the absence of a genu or body.

Agenesis of the corpus callosum
Cardinal features of corpus callosum agensis
Hypogensis of the corpus typically produces an intact genu and body with absent splenium and rostrum.
Other patterns suggest secondary destructive process

Associated anomalies, corpus callosum agenesis?
Dandy-Walker
Disorders of neuronal migration, organisation
Encephaloceles
Symptoms of callosal agenesis
Seizures, mental retardation.
Commonly also related to associated brain abnormalities
Aicardi’s syndrome

X-linked disorder
Infantile spasms, callosal agenesis or hypogensis, chorioretinopathy
Abnormal EEG

Cranium bifidum
Defect of neural tube closure
Cranial defect with herniation of meniniges (meningoceole) or meninges and brain (meningoencephalocoele) or meninges, brain and ventricles (meningohydroencephalocoele)
Varies from no functional impairment to severe motor and mental impairment with seizures
Lissencephaly
Absence of cortical gyri
Mental retardation and hypotonia or spasticity
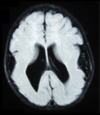

Lissencephaly
Polymicrogyria
Overabundant, undersized, cortical gyri
Mental retradation and hypotonia

Pathophysiology of intracranial lipomas
Mesenchyme gives rise to the leptomeninges
Abnormal differentiation of this mesenchyme may lead to the formation and deposition of fat in subarachnoid space
Location of intracranial lipomas from most to least common
Deep interhemispheric fissure
Quadrigeminal plate cistern
Interpeduncular cistern
CPA
Sylvian cistern
Interhemispheric lipomas
Also known as lipomas of the corpus callosum, associated with hypogensis or agenesis of the corpus callosum.
May also be evidence of punctate or curvilinear midline calcifications or the presence of other anomalies such as encephaloceles and cutaneous lipomas

Interhemispheric lipoma

Quadrigeminal plate lipoma

Interpeduncular lipoma
Def: Cephalocoele
Skull base or calvarial defect assocaited with herniaton of intracranial defects
Aetiology of cephalocoeles
Skull base cephalocoeles are defects of enchondral bone- defects in induction of bone or disunion of basilar ossification
Calvarial- defects of membraneous bone either caused by defect of bone induction or mass effect and pressure erosion by an expanding intracranial lesion or failure of neural tube closure.
Most common locations of cepalocoeles
Occipital
Frontoemthmoidal
Parietal
Nasopharyngeal
Associations with occipital cephalocoele
Callosal anomalies
Anomalies of neuronal migration
Chiari malformation
Dandy-Walker
Poor Px include HCP, microcephaly, meningoencephalocoele
Which type of cephaloceole has least favourable prognosis
Occiptal
Frontoethmoidal cephalocole
Results from failure in normal regression of dura that extends from cranial cavity to the skin through persistent foramen caecum or fonticulus frontalis
Persistence may give a dermal sinus tract which can give rise to dermoid or epidermoid tumour
Examination reveals superficial skin-covered mass or nasal dimple with hypertelorism
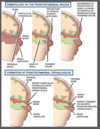

Frontoethmoidal cephalocoele
Subtypes of frontoethmoidal cephalocoele
Frontnasal ephalocle
Frontoethmoidal cephalocoele
naso-orbital cephalocele

Parietal cephalocel
Uncommoon
Poor prognosis as commonly associated with major anomalies including Dandy-Walker, callosal agensis, Chiari II and holoprosencephaly
SSS involvement is common
Atretic cephaloclee
Small, hairless, midline masses associated with sharply marginated calvarial defect and high incidence of midline anomalies e.g. porencephalies, interhemispheric cysts and callosal agenesis
Nasopharyngeal cephalocele
Very uncommon
Occult, often not presenting until 10y/o where patient presents with nasal sutffiness of excessive mouth breathing
O/E: Nasopharyngeal mass that increases with Valsalva.
Associated with callosal agensis and may include tethering of hypothalmaus and optic chiasm resulting in endocrine and visual dysfunction

Dermal sinus
3-5th week
Defect in separation of neuroectodem from surface ectoderm
Sinus commonly contains elements of both dermal and epidermal tissue
Midline found anyway between nasion and coccyx but commonly between the glabella andanion
May have associated cysts along tract


Dermal sinus
Clinical presentation of dermal sinus
Benign cutaneous cosmetic blemish to serious intracranial infection or tumourlike process due to mass effect from dermoid or epidermoid cyst
May have associated angiomata, abnormalities of pigmentation, hypertrichosis, abnormal hair pattern, subcutaneous lipomata, skin tags
Def: Arachnoid cysts
CSF containing lesions covered by membranes that consist of arachnoid cells and collagen fibres that are continuous with the surrounding arachnoid.
Pathophysiology of arachnoid cysts
Anomalous splitting and duplication of endomeninx which normally forms a loose extracellular substance in the future subarachnoid space.

Common locations of arachnoid cysts
2/3rds supratentorial most commonly Sylvian cistern, others include suprasellar, interhemispheric, intraventricular
1/3rds infraentroial- divdied between CPA, posterior to vermis and superior to quadirgeminal plate
Presentaiton of arachnoid cysts
Grading of middle cranial fossa arachnoid cysts
Galassi classification
Galassi 1
Small,spindle shaped
Limited to the anterior portion of the middle cranial fossa below sphenoid ridge
Free communication with subarachnoid space

Galassi 2
Superior extent along sylvian fissure
Displacement of temporal lobe
Slow communication with subarachnoid space

Galassi 3
Large
Fills whole middle cranial fossa
Dispalcement of temporal, frontal and parietal lobes
Often results in MLS
Little communication with subarachnoid space

What is the exception to the inside out pattern of neuronal migration
The neurones that form the most superficial layer- the molecular layer which seem to migrate first
Anomalies as a result of defect in neuronal migration
Tend to cause malformations in which cortical neurones are residing in abnormal locations or patterns.
Lissencephaly
Heterotopia
Polymicrogyria
Schizencephaly
Lissencephaly
Associated with severe mental retardation
Defective migration of cerebral neurones results in failure of cortical gyri to develop.
Cerebral hemispheres are smooth with absent cortical sulci and cerebral fissures are shallow.
Microscopically aberrant cortical cell layers
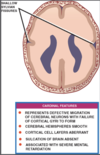
Lissencephaly etymology
Derives from greek lissos- “smooth”
Heterotopia
Collections of normal cortical neurones that fail to reach the cortex as a defect in radial neuronal migration.
May occur in isolcation or in association with other brain anoamlies.
Subtypes based on location and pattern of organisation
Heterotopia
Heter- other
Topia- place
Subtypes of heterotopia
Subependymal heterotopia
Focal subcortical heterotopia
Band heterotopia
Normal development, normal motor function.
Onset of seizures in second decade of life

Subependymal heterotopia
Dependant on size can present with normal to severely abnormal developmental delay
Motor disturbances in association with seizure disorders

Focal subcortical heterotopia

Moderate to severe developmental delay
Medically intractable seizures

Band heterotopia (AKA diffuse gray matter heterotopia)

Polymicrogyria
Varaible presentation with severe motor and intellectual dysfunction dependent on the extent of cortical involvement.
Abnormal neuronal representation resulting in distribution of neuroens into abnormal multiple small gyri
May affect variable portion of cortex in one or both areas
Most common site- posterior Sylvian fissure

With what is this abnormality closesly associated?

Polymicrogyria
Congential CMV infection
Schizencephaly
Seizures, hemiparesis, variable developmental delay determined by location, extent and number of clefts
Abnormal development of gray matter-lined cleft within the cerebral hemisphere that may extend for part of the entir distance from pia of cortex to ependyma of lateral ventricle
Can be classified as open or close lip dependent on extension into the ventricle.
Clefts comprise cortical neurones with abnormal lamination.
Most frequently occur in the region of pre/post-central gyri and can be unilateral or bilateral
Bilateral associated with worse Px


Open lip schizencephaly

Closed lip schizencephaly
Holoprosencephaly etymology
Holo- whole
Prosencelphaly- forebrain
I.e. condition of whole or inappropriately divided forebrain
Holoprosencephaly
Group of related disorders characterised by common failure of differentiation and cleavage of the prosencephalon
Three subtypes of holoprosencephaly
Alobar
Semilobar
Lobar
Outline differentiation and cleavage of hte prosencephalon
D32, germinal matrix cleaves into superior and inferior portions
Superior-> telencephalon
Inferior-> diencephalon
D32-34 lamina terminalis differenitates into the interhemsipheric cerebral commissures with associated evagination and separation of the cerebral hemispheres.
Defect in this cleavage process results in failure of transverse differentaition and cleavage into telencephalon and diencephalon and failure of the lateral differentiation and cleavage of the prosencephalon into two cerebral hemispheres
Alobar holoprosencephaly
Most severe form
Fused thalami with absent third ventricle
No interhemispheric fissure, falx or corpus callosum
Holoventricle contiugous with large dorsal cyst with only small rim of brain anteriorly
Associated with severe midline facial deformities, hypotelorism manifested in most severe form by cyclopia
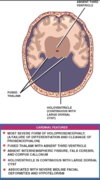

Alobar holoprosencephaly
Fused thalami with absent third
No interhemispheric fissure, falx, corpus callosum
Holoventricle continuous with large dorsal cyst
Semilobar holoprosencephaly
At least partial separation of thalami
Small third ventricle
Partially formed or absent interhemispheric fissure and falx
In contrast to the normal hypogenetic corpus in which there is a normal genu and body, holoprosencephaly is the one exception which demonstrates an intact splenium but small or absent genu and body


Semilobar holoprosencephaly
Partial separation of thalami with small third
Partially formed or absent interhemispheric fissure and falx
Intact splenium of corpus calosum (small or absent genu and body)

Lobar holoprosencephaly
Fully formed third ventricle
Intact corpus callosum
Absent septum pellucidum
Frontal lobe hypoplasia


Lobar holoprosencephaly
Mildest form
Septum pellucidum absent and frontal lobe hypoplasia
Third ventricle and corpus callosum intact

Septo-optic dysplasia
Two primary features:
Hypoplasia of optic nerves
Hypoplasia or absence of septum pellicdum
Manifests as visual distrubances including nystagmus, loss of acutity, endocrine abnormalities typically involving GH and TSH


Septo-optic dysplasia
Chiari I
Caudal extension of cerebellar tonsils below FM
May be asymptomatic or related to symptoms secondary to syringomyelia e.g. CN palsies, dissociated sensory loss.
Headache, neck and arm pain may develop in the adult


Chiari I malformation
Herniation of cerebellar tonsils below level of FM
May have associated syringomeyelia

Chiari II
Complex malformation invartiably associated with myelomeningocoele and multiple other brain anomalies
May be expained by development of a normal sized cerebellum but abnormally sized post fossa with a low tentorial attachment
Infants require repair of myelomenginocoele and shunting of HCP
May have life threatening bulbar symptoms.
Apnoea, bradycardia and nystagmus is a common clinical finding
Progressive spastic weakness and appendicular ataxia may gradually .

Cardinal features of Chiari II
Inferiorly displced vermis and multiple other anomalies
Associated with lacunar skull
Myelomeningocoele (100%)
Syringomyelia (50-90%)
HCP (90%)
Brain anomalies associated with Chiari II
Heterotopias
Polymicrogyria
Interdigitated Gyri
Partial callosal agesnesis
Large massa intermedia
Beaked tectum
Towering cerebellum with anterior creep around brainstem
Inferiorly displaced vermis
Medullary kinking


Chiari 2

Lacunar skull
Seen in Chiari 2 malformation
There is a ventricular shunt system in place
haracterised by groups of round, oval or finger-shaped pits on the inner surface of the vault (membranous part), separated by ridges of bone. They lie in the thickest part of the frontal, parietal, and upper occipital bones.
Different to copper beaten skull whcih is due to pressure, this is through defective ossificaiton

Copper beaten skull
HCP
Chiari III
Herniation of posterior fossa contents through a defect at C1-2 level (low occipital/high cervical (en)cephalocoele)
Syrinx
Rarely compatible with life

Who was Chiari?
Hans Chiari, Austrian pathologist

Chiari 3
Dandy Walker Malformation
Enlarged posterior fossa with eleveated tentorial attachment and high transverse sinus and lamboid/torcula inversion
Superior medullary velum fails to develop with foramen Magendia and Luschke atresia.
hypo or agenesis of the vermis and cystic dilatation of the fouth ventricle
Frequently accompanied by HCP (90%) of patients at time of diagnosis)
Most commonly associated with callosal agensis.
May present with developmental delay, enlarged head circumference, signs and symptoms of HCP

DDx for Dandy Walker malformation
Dandy-Walker
Mega cisterna magna
Dandy-Walker variant
Post fossa arachnoid cyst
Difference between Dandy-Walker Malformation and mega cisterna magna
Presence of communication between the fourth and the cisterna magna

Mega cisterna magna
Difference between Dandy-Walker and Dandy Walker variant
Dandy walker variant is characterised by hypogenesis of the cerebellar veris and cystic dilatation of the fourth with a normal sized posterior fossa
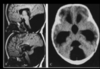
Dandy Walker

Dandy-Walker variant
Dandy Walker Variant
Cerebellar vermis hypoplasia
Vallecular enlarged- open communication between 4th and cisterna magna
Normal posterior fossa
HCP in 25%
Mega cisterna magna
Large cisterna magna with CSF accumulation in post fossa
Normal 4th ventricle with no HCP
Fills with intrathecal contrast
Post fossa arachnoid cyst
Posterior fossa CSF accuulation with normal cerebellum and 4th
May or may not be associated with HCP
Cyst does not fill with intrathecal conrast (unlike mega cisterna magna)
Absence of vermis
Enlarged fourth
Fills with intrathecal contrast
HCP in 85%
DWM
Vermian hypoplasia
Enlarged 4th
FIlls with intrathecal contrast
HCP in 25%
DW variant
Normal cerebellum
Normal 4th ventricle
Enlarged cisterna magna
Fills with intrathecal contrast
No HCP
Mega cisterna magna
Normal cerebellum
Enlarged cisterna magna
Normal fourth
Doesn’t fill with intrathecal contrast
+/- HCP
Post fossa arachnoid cyst
Lhernitte-Duclos Syndrome
AKA diffuse hypertrophy of cerebellar cortex/dysplastic cerebellar gangliocytoma
Histologically cortex demonstrates a thick layer of abnormal ganglion cells that occupy the granulary, thick hypermyelinated marginal layer and thin Purkinje layer
Disorder may extend into the vermis or the contralateral hemisphere
Mass effect may produce cerebellar symptoms but many affected individuals are asymptomatic


Lhermitte-Duclos disease
Common defect in phakomatoses
Share a common defect in the development of ectodermal structures including nervous, structures, skin, retina.
4 major phakomatoses
NF
TS
Sturge-Weber
VHL
Major intracranial lesions associated with NF1
Optic glioma
Cerebral astrocytomas
Sphenoid wing dysplasia
Plexiform neurifbromas
Bourneville’s disease
Tuberous sclerosis
Tuberous Sclerosis
Triad of mental retardation, epilepsy and adenoma sebaceum though only present in half of patients
Predominantly consists of hamartomatous lesions involving brain, eye, visceral organs
Cutaneous manifestations of tuberous sclerosis
ADenoma sebaceum- adenoma involving face
Ash leaf macules- depigmented naevi involving trunk and extrimities
Shagreen patches

Adenoma sebaceum
?Tuberous sclerosis

Ashleaf macules ?tuberous sclerosis

Shagreen patch
Cranial lesions associated with tuberosu sclerosis
Subependymal hamartomas
Giant cell tumours
Cortical tubers
WM lesions
Systemic lesions associated with tuberous sclerosis
Hamartomatous lesions involving kidneys- angiomyolipoma
Heart- rhabdomyoma
Lungs- lymphangiomyoma
Eye- retinal hamartoma

Tuberous sclerosis: T1-weighted FLAIR images reveal cerebral hamartomas as low signal intensity subcortical lesions, subependymal hamartomas casted on the ventricular space and small giant cell astrocytomas adjacent to the foramen of Monro.

Cortical tubers
Most common brain lesion associated with tuberous sclerosis
Subepednymal hamartomas
How to differentiate between subependymal hamartomas and subependymal giant cell tumours
Giant cell tumours tend to have intense contrast enhancement on MR
Are larger
Tend to more frequently occur near the foramen of Monro which can cause non-communicating HCP

Cortical tubers
Characteristic hamartomas mae of bizarre giant cells, fibrillary gliosis and siorderd myelin sheaths which appear as smooht, slightly raised subcortical nodules
Sturge Weber Syndrome
ANgiomatosis involving face, choroid of eye and leptomengines
Facial angioma follows the opthalmic division of V.
Localised atrophy and caclification of the cerebral cortex ipsilateral to facial lesion is characteristic.

Retinal hamartoma

Sturge Weber Syndrome
Clinical presentation of Sturge Weber
Seizures
HH
Variable mentral retardation
VHL
AD characterised by
CNS:
retinal angiomas, cerebellar and spinal cord haemangiobolastomas
Systemic:
RCC, phaeo, angiomas of liver and kidney and pancreatic, kidney, liver, epidiymal cysts
Genetic basis for VHL
Autosomal dominant on chromosome 3

Retinal angioma
Most common location of spina bifida occulta
L5 and S1 vertebral arches
Spina bifida oculta
Radiological diagnosis without external signs of developmental anomlay
Characterised by absence of one or more spinous process with associated vertebral arch hypogenesis
20-30% incidence in general population
Meningocoele
Ctystic skin or membrane covered mass consisting of a meningeal sac containing onle CSF that is continuous with the CSF of the spinal canal.
Pathophysiology of meningocoele
Aetiology unclear, malformation appears to represent a postneurulation defect developing after the normal disjunction of neuroectoderm from cutaneous ectoderm.
Thought to result from defect in mesenchymal and cutaneous ectodermal development.
Myelomenginoceole is a true defect in neurulation
Pathophysiology of myelomeningocoele
Defect in disjunction where the neuroectoderm and cutaneous ectoderm separatae
Results in neural placode made of cells that would normally form ependymal lining of neural tube.
Because the placode remains attached to the skin, the mesenchymal elements are unable to migrate and fuse leading to several vertebral anomalies.
Vertebral anomalies in myelomeningocoele
Absence of spinous processes and laminae
Reduction in AP size of vertebral bodes
Increased interpedicular distance
Large laterally extending transverse processes
May contribute to kyphoscoliotic deformities in 1/3rd
Sequelae from myelomeningocoele
Chiari (100%)
HCP
Tethered SC
Myelomenginoceole avove L3
Complete paraplegia
Dermatomal para-anaesthesia
Bladder/rectal incontinence
Nonambulatory
Myelomeningocoele L4 and below
Manifestation as for L3 except preservation of hip flexors, abductors and knee extensors
Ambulatory with aids, bracing and orthopaedic surgery
Myelomeningocoele S1 and below
As for L4 except with preservation of feet dosriflexors and partial preservation of hip extensors and knee flexors
Ambulatory with minimal aids
Myelomeningoceole S3 and below
Normal lower extremity motor function
Saddle anaesthesia
Varaible bladder/rectal incontinence
Spinal lipoma
Skin-covered dorsal masses of fat and connective tissue in continuity with leptomeninges or spinal cord
Result of premature disjunction which results in migration of mesenchymal tissue into the ependymal lined central canal of the neural tube, differenitates into fat due to connection with central canal of neural tube

Categories of spinal lipoma
Intradural (4%)
Lipomyelomeningocoeles (84%)
Fibrolipomas of filum terminale (12%)
Intradural lipomas
Commonly occur in thoracic region
PResent with signs and symptoms of cord compression in the adult

T1 saggital MR thoracic spine
Intradural thoracic lipoma
Fibrolipomas of filum terminale
Take origin from an abnormality in caudal cell mass during secondary neurulation
Asymptomatic but may present with symptoms of tethered cord


Fibrolipoma of the filum terminale
Lipomyelomeningocoeles
Present as subcutaneous, skin covered lumbosacral masses.
Most common form of spinal lipomas
Extend beyond the dorsal surface of the neural placode through spina bifida.
At the level of the lipoma, the dura is deficient in the dorsal midline, allowing free medial edges to attach to the neural placode which is extradural as a consequence

Describe the orientation of the neural placode in lipomyelomeningoceoele
As the neural placode herniates through the bony spina bifida, it rotates.
This places the dorsal surface of the neura placode laterally or dorsolaterally rather than straight dorsally.
The nerve roots on each side assume asymmetric lengths such that those on superficial side grow longer than those on deep.
The shorter roots may result in inferior tethering of the spinal cord
Differnce between myelomenginocoele and lipomyelomeningocoele
Lipomyelomengincoele is skin covered, marked by a lipoma attached to the dorsal surface of the placode

Lipomyelomeningoceoele
Common associations with lipomyelomeningoceole
Orthopaedic foot deformities
Sacral anomalies
Segmentation anomalies
Anatomical definitiion of split cord
Fissure separating spinal cord >=1 segements +/- bony spicule/ fibrocartilaginous septum from dorsul VB
Each hemicord contains own set of roots.
May have separate or shared dural sleeve which predicts absence of bony septum
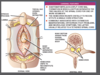
Type 1 diastomatomyelia
duplicated dural sac
hydromyelia common
midline spur often present (osseous or osteocartilaginous)
vertebral abnormalities: hemivertebrae, butterfly vertebrae, spina bifida, fusion of laminae of adjacent levels
skin pigmentation, haemangioma and hypertrichosis (hair patch) are common
patients are usually symptomatic presenting with scoliosis and tethered cord syndrome
Diastematomyelia type:
duplicated dural sac
hydromyelia common
midline spur often present (osseous or osteocartilaginous)
vertebral abnormalities: hemivertebrae, butterfly vertebrae, spina bifida, fusion of laminae of adjacent levels
skin pigmentation, haemangioma and hypertrichosis (hair patch) are common
patients are usually symptomatic presenting with scoliosis and tethered cord syndrome
Type 1
Type 2 diastomatomyelia
single dural sac and no spur/septum
cord divided, sometimes incompletely so
hydromyelia may be present
spina bifida may be present, but other vertebral anomalies are far less common
patients a less symptomatic or may even be asymptomatic
Diastomatomyelia type
single dural sac and no spur/septum
cord divided, sometimes incompletely so
hydromyelia may be present
spina bifida may be present, but other vertebral anomalies are far less common
patients a less symptomatic or may even be asymptomatic
Type 2

Diastomatomyelia Type 1

Diastomatomyelia Type 2
Assocoations with diastomatomyelia
Cutaneous manifesations- naevia, hypertrichosis, lipomas, dimples, haemangiomas in >50%
Orthopaedic foot problems
Neurological symptoms 2o to cord tethering
Caudal agensis
Group of caudal malformations demonstrating partial or complete absence of either or both lumbar and sacral vertebra with absence of corresponding neural tube.
Can include hemivertebrae, wedge shaped vertebrae, fused vertebrae, sacralisation of lumbar vertebraie
Distal SC absent and terminus of remaining cord ends in dyspplastic glial nodule.
Motor deficits correspond to level of lesion, sensory defects may be absent due to sparing of neural crest cells.
50% have myelomeningocoeles
Associations with caudal agenesis
Limb malformations- flattend buttocks, gluteal atrophy, equinvoarous defomities
Visceral malforamtions: Tracheo-oesophageal fistula, Meckel’s diverticulum, cloacal estrophy, omphalocoele, malrotation, renal agenesis, horeshoe kidney, uretral and bladder duplications, anomalies of external genitalia


Caudal agenesis
