Key notes final Flashcards
(78 cards)
What do computer simulations do and what do they allows?
Allow the study of properties of a many body system by creating a mathematical model (e.g a molecular mechanics forcefield) to describe the system behaviour which can be run to sample configurational space of that system (main families are MS and MC)
Why bother using comupter simulations?
- Gain better understanding of the system at a molecular level which allows better design of experiments and help interpret data.
- Predict properties/behaviour from molecular structure.
- Simulate unrealistic experiments that explore extreme conditions or toy models (e.g turn off charges to investigate hydrophobic driven reactions)
What is an MD simulation and what does it allow us to do?
- Enable us to simulate the time evolution of a system of particles.
- Can extract thermodynamic and kinetic information through simulation of particles at a position at the beginning and end of many steps.
- Trajectories are generated through classical dynamics (ignore electronic effects) by solving newtons equations of motion.
How are newtonian principles employed in MD simulations?
- The force acting on an atom i due to all other particles in a system is linked to the potential experienced by atom I and its position
- Using newtons second law (F=ma) we can also find the particles acceleration
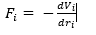
Why can MD trajectories not be solved analytically? What must be done instead
- Cannot be solved analytically for a many-body system as force acting on each particle depends on the position of all other particles at that moment in time.
- Instead F=ma is solved numerically using small time steps Δt (~fs) where the force is calculated at the end of each of these time steps then following on to the next one.
- Most commonly done with the Verlet algorithm
What defines the transferability of a force-field?
- The ability to re-use fore-field parameters across a range of different functional groups.
- More parameters may be accurate for a specific system but transfer poorly to another.
- Forcefields have been paramaterised to reproduce properties of proteins, lipids e.g CHARMM, AMBER. Same functional form but differing parameters (different torsion angle for amino acids)
Ergodicity is not always true in practice, why is this?
- A barrier that is too high can cause system to be kinetically trapped in a certain region of phase space, leaving the global minimum unexplored.
- If one is doing a grid-based lattice simulation getting stuck in local minima when starting on certain set of lattice coordinates can be a problem.
What are periodic boundary conditions (PBC)?
Convenient way of representing bulk solution without having to simulate every molecule where molecules are placed in a simulation cell replicated in all three directions via minimum image convention
What is the time scale problem? What is a chemcial example that suffers from this?
- Larger than that of length scale, avoiding getting stuck in local minima can use up most simulation time without sampling desired configurational space.
- Can occur if waiting for a rare event to occur, but instead getting stuck behind a kinetic barrier.
- No way to know if this has occurred as the PES is not available prior to the simulation.

What are edge effects and how are they overcome?
- Many small boxes have many atoms, some of which are close to edge.
- Results will be influenced by particles interacting with the edge of the system.
- Edge effects are overcome by having molecules that pass through the cell walls appear on the other side.
Assuming MM suffices over QM, what is the length scale problem?
- System must be a manageable number of particles; however systems are growing larger (millions of atoms).
- One must choose the system carefully to get desired output.
Briefly discuss and compare three different force-fields, using the alcohol octanol as an example.
- All-atom: An all atom/atomistic force-field treats each atom explicitly. At every step, the simulation must input all bonding/non-bonding interactions in a system. In long molecules like octanol, this can lead to many dihedral angles needing to be specified for all differing ~-OH chains. Very precise however, computationally costly.
- United-atom: cheaper way to describe the system that saves computational time, but still expensive. It does this by grouping less chemically involved hydrogen atoms into CH2 groups. Good for lipid descriptions
- Coarse graining: 4-5 heavy atoms with their H’s are grouped into a single interaction site, saving large amount of computational effort. Octanol is split into a polar (hydrophilic) and non-polar (hydrophobic) half. Lose a degree of chemical detail as cannot capture hydrogen bonding explicitly. However partial charges assigned to these blocks to describe electrostatic and non-bonding interactions.

What are three ways in which coarse graining allow simualtion on larger systems for longer timescales?
- Decrease in total number of particles N
- Larger time step
- Smoother energy landscape
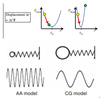
How does larger time-step ‘speed up’ a coarse grain simulation?
- A time step for integration needs to be smaller than the highest frequency motion in a system.
- The verlet algorithm can be used to move an atom through calculation of the force, with respect to all other atoms, to apply in one timestep, which should be constant.
- A time step may be suitable for bond stretch of a low frequency (left), resulting in it being closer to its equilibrium value. However, the same force acting on a higher frequency motion (right) results in a high energy final position, leading to assumption of force being constant over our chosen timestep to be invalid.
- Timesteps should be 10x smaller than highest frequency motion, which are generally bond stretches of light hydrogen atoms.
- By removing bonds involving them, a larger time step can be used, as particles of higher mass oscillate slower. Each time step now propagates us further forward in time.
How does a decrease in total number of particles N ‘speed up’ a coarse grain simulation?
- A decrease in total number of particles reduces the number of force calculations required at each step.
- Resulting in less computational time needed to move the system forward one-time step, as less forces and energies must be calculated each step.
How does a smoother energy landscape ‘speed up’ coarse graining?
- Rugged energy landscape of an atomistic model has many local minima.
- Averaging that occurs through coarse-graining results in a smoother energy landscape with less local trapping/friction and faster dynamics.
- Beads can slide over each other much easier
Why must we develop new interaction potentials for coarse grained systems?
- Coarse-grained particles do not interact in the same way that atoms do.
- New coarse-grained potentials must be developed that are unique to each system.
- Parameterising is a large challenge as takes a lot of time to predict the correct output.
What are two methods of generating course-grained interaction potentials?
- Use an analytical functional form with associated parameters; like atomistic interaction potentials but with CG beads
- Numerically fitting data from atomistic simulations using tabulated potentials (list of r’s between atoms with corresponding F’s/E’s – useful for complex systems where don’t have analytical function)
What is mapping, and why is it essential for developing a CGP? (PPQ)
- The first stage in developing a CG interaction potential (CGP) is to define mapping of the atomistic system in to a CG one.
- Essential for new molecules and there are many ways to assign atomistic structure depending on what interactions are expected to be the focus.
What is an RDF?
- A radial distribution function g(r) shows the probability of finding a particle at distance r from a reference particle.
- At small distances there is a low probability of finding another atom due to coulombic repulsion (LJP?)
- Shells form around the reference particle as other bands of particles appear, with depletion zones between them.
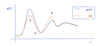
What are 2 general rules of mapping?
- Each bead will inherit the mass/charge of its constituents
- Must maintain the overall shape of the molecule (otherwise may not replicate the correct self-assembled structure)
Outline how you would produce a CG trajectory from an atomistic simulation using a bottom up approach
- First, run a reference atomistic simulation to use as our benchmark (however does not necessarily equal experimental/ab initio PES)
- Extract effective CG potentials using a trajectory of a system (a series of snapshots over time).
- Each snapshot in the atomistic simulation will have positions for all atoms at each timestep which will be used to find their corresponding Force/Energy.
- A CG pseudo trajectory can be attained by grouping atoms in to beads using the beads centre of mass based on an atomistic average.
- How does our improved guess for the intermolecular CG potential [VCGi+1(r)] change when gi(r) = gref(r)? – 1 on graph
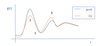
kBTln…= 0, no change to interaction potential needed as CG simulation produces same pairwise structure as atomistic

- How does our improved guess for the intermolecular CG potential [VCGi+1(r)] change when gi(r) ref(r)? – 2 on graph
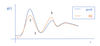
- kBT… is -ve
- probability of finding particle (correlation between CG beads) at that r underestimated.
- Equation will lower pairwise CG energy at that r, making interaction more favourable