Key notes Flashcards
- How is the ESE approximated further?
- ESE still too complex to solve e.g. due to 2nd differential in KE term
- Separate into a universal (Fe) and an external potential (Vext)

- (IMP) Outline briefly how the ESE is solved ab-intio via wavefunction theory
- (DFT is also ab initio just different appraoch)
- Exact ESE is solved through approximation of the ψ through variational or perturbation theory
- Uses HF of non-interacting electrons in a mean field
- Electron correlation captured through post HF methods
- E.g. CI, MP2 only differing in how they approximate ψ
- (IMP) Why might it be more appropriate to use electron density, ρ(r) to solve the ESE
- Ψ depends on 3n coordinates for n electrons
- Storing this info for >100 electrons is unfeasible
- ρ(r) depends only on 3 coordinate (x,y,z), describing the probability distribution of electrons in space at that point
- much simpler as only depends on position you measure it.
- What is the problem with expressing the universal function F in terms of density?
- Expansion of the original KE term contains a 2nd derivative of ψ which cannot be formulated as a simple function of ρ
- We know that F[ρ] must exist but can’t write it down.
- (IMP) Describe the details of this figure
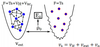
- Adiabatic connection of an artificial auxiliary system, vs and a real many-body system, vext connected by the same electron density, ρ
- Real system defined by coulomb interaction of all particles (complex)
- Mapped into a non-interacting system encoded by overall potential describing indirect interaction between atoms.
(IMP) Describe the key differences between Wave function and KS theory
- Wave function theory
- Complexity in high dimensional ψ via exact ESE
- Finite sum over slater determinants
- Finite approximation of ψ àMP2,CCSD etc
- Very slow convergence to get exact Ecorr but results in exact path via systematic improvements through variational theory
- Very computationally expensive (limited to ~30 atoms)
- DFT
- Complexity in vXC (less as based on density alone)
- FInite sum over KS equations describing orbital like states
- Non-local potential in space (and time) connects all possible interactions
- Local approximations to vXC makes systematic path to true answer difficult
- Good error calculations
- Computationally very efficient
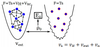
- Every external potential can have only … density
- But the same density can connect to … … external potentials
- Therefore, we can calculate the GS density of …-… e-s (via Hartree theory) in an … … vs that is known
- Use this … … potentials equivalent … to calculate the energy of the system described by …
- Every external potential can have only one density
- But the same density can connect to two different external potentials
- Therefore, we can calculate the GS density of non-interacting e-s (via Hartree theory) in an effective potential vs that is known
- Use this made up potentials equivalent ρ to calculate the energy of the system described by vext
- What is the basic assumption of Local-density approximations (LDA)?
- EXC only depends on the value of the local electron density at one point
- Discuss some other problems with LDA’s
- Binding energies are too negative i.e.overbinding
- Activation energies are unreliable
- Band gaps, ionization energies and electron affinities are strongly underestimated.
- What are Generalized-gradient approximation (GGA’s) and how do they improve upon LDAs
- Similar form but a local gradient of electron density is included as well as the value of density
- This is to gain information on the local variation in neighbouring electron density at other positions

- How does the bulk lattice constants and cohesive energies with GGA compare with LDA?
- Bulk lattice constants (unit cell vecotr between atoms): GGA increase due to more repulsive core-valence XC
- Cohesive energies (E released by binding atoms in to a solid): GGA reduction mostly due to valence effect, giving better description.
- How do Energy barriers of GGAs compare to LDAs
- Free energy of molecule better described with GGAs, reducing the degree to which the barrier is underestimated
- LDAs underestimate to a large extent.
Name an improvement GGAs make on LDAs
- GGA correct LDA overbinding, with less stiffness of tightly packed system
- Where do GGAs still fall short
- Still no long-range description of vdW forces as local approximation (same as LDA)
- As GGA favours low coordination (large gradient), can now interpret different E sites on a surface (LDA could not distinguish). However can do so incorrectly.
- What is the general idea of meta-GGAs?
- Expand the local function dependence to occupied KS orbitals.
- 2nd derivative of KE added via T[ρ] of non-interacting electrons i.e. the product of the derivative of KS ψ of single particle space
- This leads to the function becoming explicitly orbital dependent.
- Still technically local derivative however accounts for many variations in different orbitals.
- For their simplicity, LDA/GGAs perform well for a large range of materials, marking ‘semi-…’ DFT as an important improvement in how we describe the …
- However major failures in the … of chemical reduction barriers and … … as well as the overestimation of … mark a need for further improvement.
- For their simplicity, LDA/GGAs perform well for a large range of materials, marking ‘semi-local’ DFT as an important improvement in how we describe the EXC
- However major failures in the underestimation of chemical reduction barriers and band gaps as well as the overestimation of polarizabilities mark a need for further improvement.
(IMP) Describe the band-gap problem in GGA’s
- Underestimation of difference in ionisation energy/electron affinity due to self-interaction error
- Contrary to HF, VX and VH + VC don’t cancel for an H-atom
- Missing VX causes half electrons to move apart from each other instead of contracting in to a positive and negative ion in dissociation.
- How do meta-GGAs compare to LDA/GGA
- Better molecular binding E’s
- Better cohesive/structural description
- Band gaps still underestimated
- Long range vdW still not accounted for as still a local functional
(IMP) How does the error in delocalization affect how the ionisation energy and electron affinity are related to the energies of the KS states at this stage? Use a sketch to support your answer
- Energies of the KS auxiliary states do not equal the electron affinity and ionisation energy of the system as they should
- This is due to the presence of half electrons in local description causing a bowing of the curve instead of clear definition
- means half an electron in LUMO and HOMO instead of 1 in LUMO (I underest, A overest, E underest)
- Gradient represents energy levels and are incorrect

(IMP) How do 4th-rung hybrid functionals solve the localization problem in meta-GGAs? Use a sketch to support your answer
- Includes HF exchange, which strongly over localizes/stabilizes electrons, making graph more convex.

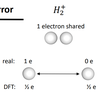
(IMP) What is the over localisation error also associated with the band gap problem?
- Electronic states are too localised as electrons do not sufficiently repel each other (Pauli repulsion missing)
- Electrons do not come close in contact unless they must (run away from themselves as orbitals more diffuse.
(IMP) Describe the problem of missing long-range correlation in local functionals
- Electron correlation VC treated incorrectly (due to local approximation)
- Leading to no long range interaction between non overlapping densities
- As a result, LDA/GGA/meta-GGA do not capture dispersion/vdW effects
(IMP) What is the solution to the long-range correlation error?
Include explicit non-local correlation or long-range dispersion terms, describing sum of all pairwise interactions as a function as 1/r^6 e.g. Many body dispersion/ vdWsurf
- How do hybrid functionals compare to local?
- Large improvements in barriers, band gaps and dissociation energies (≈0.1 eV expt)
- More accurate and consistent description of CO site adsorption.
- What is the idea of 5th rung RPA (Random Phase Approximation) functionals?
- Xc functionals become explicitly dependent on unoccupied orbitals as well as occupied orbitals previously
- Similar to CCSD in HF (mixing excitation to unoccupied states)
