Fatty Acid Metabolism Flashcards
Significance of Fatty Acids
- Most lipids contain or are derived from fatty acids.
- Structural components of:
- Triacylglycerols (TAG) aka triglycerides (TG)
- 3 FA esterified to a glycerol backbone
- Phospholipids (PL)
- 2 FA esterified to phosphorylated glycerol
- Glycolipids (GL)
- 1 FA linked to a glycosylated ceramide
- Cholesteryl ester (CE)
- 1 FA esterified to steroid ring of cholesterol
- Triacylglycerols (TAG) aka triglycerides (TG)
- Precursors in the synthesis of eicosanoids
- Prostaglandins
- Thromboxanes
- Leukotrienes
- Synthesized from acetyl CoA
- Catabolized to acetyl CoA
- Disturbed in a number of pathological processes

Fatty Acid Structure
- Carboxylic acid group (COO-) ⇒ C#1
- C#2 ⇒ α, C#3 ⇒ β
- Alkyl chain, typically:
- Linear (unbranched)
- Methyl (-CH3) terminus ⇒ ω-carbon
- Even # of carbons
- Long
- SCFA: 2-4 C
- MCFA: 6-12 C
- LCFA: 14-20 C
- VLCFA: ≥ 22 C
- If unsaturated:
- Monounsaturated (MUFA) ⇒ 1 C=C
- Polyunsaturated (PUFA) ⇒ 2 or more C=C
- In cis configuration
- At 3 carbon intervals
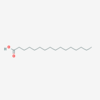
Palmitic Acid
IUPAC: Hexadecanoic acid
Carboxyl-Reference: 16:0
Omega (ω) - reference: 16:0
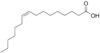
Palmitoleic Acid
IUPAC: 9-hexadecenoic acid
Carboxyl-Reference: 16:1Δ9
Omega (ω) - reference: 16:1 (ω-7) or (n-7)
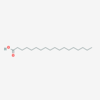
Stearic Acid
IUPAC: octadecanoic acid
Carboxyl-Reference: 18:0
Omega (ω) - reference: 18:0
Oleic Acid
IUPAC: 9-octadecenoic acid
Carboxyl-Reference: 18:1∆9
Omega (ω) - reference: 18:1 (ω - 9) or (n - 9)

Linoleic Acid
IUPAC: 9,12-octadecadienoic acid
Carboxyl-Reference: 18:2∆9,12
Omega (ω) - reference: 18:2 (ω - 6) or (n - 6)
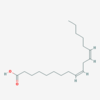
α-linolenic Acid
IUPAC: 9,12,15-octadecatrienoic acid
Carboxyl-Reference: 18:3∆9,12,15
Omega (ω) - reference: 18:3 (ω - 3) or (n - 3)

Arachidonic Acid
IUPAC: 5,8,11,14-eicosatetraenoic acid
Carboxyl-Reference: 20:4∆5,8,11,14
Omega (ω) - reference: 20:4 (ω - 6) or (n - 6)
Fatty Acid Synthesis Overview
In humans the overall reaction is:
8 Acetyl Co-A + 7 ATP + 14 (NADPH+H+)
→
Palmitate (16:0) + 8 CoA + 7 (ADP + PI) + 14 NADP+ + 6 H2O
- Cytosolic process especially important in the liver, CNS, lactating mammary gland, and adipose tissue
- Endergonic and reductive
- Acetyl-CoA from glycolysis and PDH reaction
- NADPH from pentose phosphate pathway and malic enzyme
- Catalyzed by two enzymes
Citrate Shuttle
Acetyl CoA is generated in the mitochondrial matrix (by glycolysis and PDH complex) but fatty acid synthesis occurs in the cytosol.
CoA cannot cross the inner mitochondrial membrane.
Acetate transported out as citrate without the crossing of CoA.
-
Acetyl CoA is combined with oxaloacetate in the mitochondrial matrix to form citrate by citrate synthase with the release of CoA.
- Glucose breakdown inhibited in the liver during times of excess energy ⇒ High [ATP] inhibits Isocitrate dehydrogenase of TCA cycle. Isocitrate easily interconverted to citrate.
- Citrate transported out of the mitochondria to the cytosol where it is converted by citrate lyase + CoA into Citrate-CoA.
-
Citrate CoA cleaved by citrate cleavage enzyme with use of 1 ATP back into Acetyl CoA and Oxaloacetate.
- Enzyme positively regulated in response to insulin.
- Acetyl CoA used in FA synthesis.
- Oxaloacetate converted to Malate by Malate Dehydrogenase with the use of 1 NADH.
- Malate can either:
- Return to the mitochondria via the malate shuttle.
- Be converted in the matrix back to oxaloacetate by malate dehydrogenase with the production of 1 NADH.
- Be converted in the cytosol to Pyruvate by Malic Enzyme releasing CO2 and producing 1 NADPH
- Another source for NADPH used in FA synthesis
- Return to the mitochondria via the malate shuttle.

Coenzyme A
- Often-used carrier of activated acyl groups
- Acetyl
- Fatty acyl
- Others
- Thioester linkage has a large negative ΔG°’ of hydrolysis of -7.5 kcal/mol
-
Phosphopantetheine group acts as a long arm which shuttles substrates similar to E2 of PDH & α-KGD complexes
- Contains pantothenic acid (Vit B5)
- Not synthesized in humans so it is an essential nutrient
- Contains pantothenic acid (Vit B5)

Fatty Acid Synthesis
Part 1
Acetyl CoA (2C) + Carbon Dioxide (form of HCO3-) + ATP
→
Malonyl CoA (3C)
Catalyzed by acetyl CoA carboxylase (ACC)
- Committed and rate-liming step of FA biosynthesis
- Irreversible reaction
- ACC affected by allosteric and covalent regulation
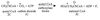
Acetyl CoA Carboxylase
(ACC)
- Rate-limiting enzyme of fatty acid biosynthesis
- Requires biotin coenzyme
- Shuttles CO2 to acceptors
- Swinging arm mechanism
-
Short-term regulation
-
Allosteric Effectors
- Pos: citrate
- Neg: end product ⇒ palmatate in humans
-
Covalent Regulation
- Activated: dephosphorylation
- Inactivated: phosphorylation
- By AMP-activated protein kinase (AMPK) or PKA
- AMPK allosterically activated in response to an increase in AMP/ATP ratio such as with hypoxia, exercise, etc.
- So when [ATP] low FA biosynthesis is not attempted because ACC requires ATP
- Citrate + dephosphorylation favors the polymerization of the protein into the active form.
-
Allosteric Effectors
-
Long-term regulation via gene expression
-
Diet controlled:
- High carb diet increases expression of ACC via trans-acting ChREBP at cis-acting ChoRE.
- High fat diet decreases transcription.
-
Hormone controlled:
- Insulin increases ACC expression via trans-acting SREBP (Sterol Regulatory Element Binding Protein)
-
Diet controlled:
Fatty Acid Synthesis
Part 2
Adds 2C’s from malonyl CoA to the carboxylate end of an acyl acceptor through a repetitive 4-step sequence:
- Condensation (decarboxylation)
- Reduction (requires NADPH)
- Dehydration
- Reduction (requires NADPH)
Catalyzed by a single homodimeric multifunctional protein
Fatty Acid Synthase (FAS)
6 catalytic activities + 1 acyl carrier protein (ACP) domain
ACP domain contains phosphopantetheine group (as seen in CoA) which acts as a swinging arm between catalytic domains.
- The initial acyl acceptor, Acetyl CoA (2C), is loaded onto the ACP domain of FAS then transferred to a cys residue in the condensing enzyme domain (CE) via the transacylase activity (#1) releasing CoA.
- Malonyl CoA (3C) previously formed by ACC is transferred to the ACP domain via the transacylase activity releasing CoA.
- Condensation reaction involves condensation of malonyl and acetate releasing the CO2 which was previously added by ACC (decarboxylation) catalyzed by the β-ketoacyl ACP synthase (KS) domain aka condensing enzyme (#2).
- First reduction of the keto-group by β-ketoacyl ACP reductase (KR) activity (#3) requiring 1 NADPH yields a hydroxyl.
- The D-isomer hydroxyl is dehydrated by β-hydroxyacyl ACP dehydratase (DH) activity (#4) to yield a trans-double bond.
- Second reduction of the trans-double bond by trans-enoyl ACP reductase (ER) activity (#5) requiring 1 NADPH yields butyryl (4C).
- Butyrl acts as the next acyl acceptor and the cycle repeats adding 2 C’s from malonyl CoA each time.
- After 8 total cycles yielding palmitate (16C) the active site is not able to accomodate anything larger and the fatty acid is cleaved from the ACP domain by the thioesterase activity (TE) (#6).
- The terminal two carbons of palmitate are from acetyl CoA and the rest are from malonyl CoA.

Fatty Acid Synthase
Regulation
- No short term regulation
- Long-term control by transcriptional regulation
- Expression increases with high carb diet
- Expression decreases with high fat diet
- Elevated [glucose]blood causes increase in [insulin] resulting in increased expression of FAS
- Also ACC, malic enzyme, and G6PD
Elongation of
Dietary and Endogenous Fatty Acids
- Occurs in the SER and mitochondria
- Mechanistically similar to de novo synthesis of FA ⇒ 2C’s from malonyl CoA added at a time to the carboxylate end
- Differences:
- More than 1 enzyme involved ⇒ elongase system
- Enzymes are mostly ER-membrane proteins
- CoA esters are used to move the substrate instead of ACP
- Substrates of ≥ 10C’s
- Product is typically 18C

Desaturation of
Dietary and Endogenous Fatty Acids
- Insertion of a C=C
- Occurs in the SER
- Fatty acid and NADH both get oxidized as O2 is reduced to 2 H2O
- Involves:
- Desaturases
- Cytochrome B5
- NADH-cytochrome B5 reductase flavo-protein (FAD containing) located in the ER membrane
- Humans have four desaturases: Δ9, Δ6, Δ5, Δ4
- Cannot insert C=C between C10 and ω-C
- Substrates primarily ≥ 18C
- 18:1Δ9 is the most common

Generation of Arachidonic Acid
Elongation Paired with Desaturation of Dietary FA
- Humans unable to make ω-6 or ω-3 unsaturated FA, however, they are dietary essential nutrients for the production of other PUFA such as arachidonic acid
- Precursor of eicosanoid hormones
- Linoleic acid (18:2Δ9,12) taken in by diet
- Undergoes Δ6 desaturation to form 18:3Δ6,9,12
- Elongated to form 20:3Δ8,11,14
- C=C are pushced back by addition of 2C to the carboxylate end
- Undergoes Δ5 desaturation to form 20:4Δ5,8,11,14 (arachidonate)
There is no interconversions between families ⇒ ω-6 family was unchanged after elongation and desaturation.

Storage of Fatty Acids
- FA stored as triglycerides/triacylglycerols (TAG)
- Composed of 3 FA esterified to glycerol
- TAG stored within a single large anhydrous droplet coated with a monolayer containing:
- Phospholipids
- Cholesterol
- Proteins such as perilipin
- More efficient form of stoerage compared to glycogen
- TAG synthesized primarily in the liver from glucose
- Sent out into the blood as VLDL
- Stored primarily in adipocytes of white adipose tissue
Retrieval of Fatty Acids
- Triglycerides stored in adipose tissue degraded to fatty acids x 3 plus glycerol by adipose lipases
-
Hormone-sensitive lipase
- Phosphorylated and activated by PKA in response to catecholamines primarily
- Phosphorylation of perilipin by PKA allows P-HSL to bind to P-perilipin on the lipid droplet
-
Hormone-sensitive lipase
- Glycerol can be used by the liver for gluconeogenesis.
- Fatty acids carried in the blood on albumin, picked up by muscle and liver, activated, and oxidized to acetyl CoA and ultimately to CO2 in mitochondria providing significants amount of ATP.
- ATP and NADH used for gluconeogenesis in liver
- Used in muscles to spare glucose for glucose-dependent tissues
- During prolonged fasting, 80% of energy is supplied by lipolysis.

Fatty Acid
Activation
- Activation involves formation of CoA esters and is required for FA participation in metabolism.
- SCFA and MCFA able to enter the matrix and activated there.
- Catalyzed by fatty acyl-CoA synthetases
- Requires ATP and CoA
- Enzyme produces PPi which is hydrolyzed to 2 Pi which drives the reaction forward.
- Family of synthetases based on chain length:
- SCFA/MCFA in mitochondrial matrix
- LCFA synthetases in the outer mitochondrial membrane but faces the cytosol, in SER, and peroxisomes.
- VLCFA in peroxisomal membrane only
Carnitine Shuttle
- LCFA are activated in the cytosol but oxidation occurs in the mitochondrial matrix.
- CoA unable to cross the inner membrane.
- FA attached to carnitine in order to facilitate transport.
- Carnitine is made from lys in the liver and kidney primarily.
For long chain fatty acids:
- FA attached to CoA by acyl CoA synthetase located on the outer mitochondrial membrane.
- Fatty acyl CoA able to cross the outer mitochondrial membrane.
- In the intermembrane space, fatty acyl group transferred from CoA to carnitine by Carnitine palmitoyl-transferase I (CPT-I) aka carnitine acyltransferase (CAT-I)
- Rate-limiting and regulated enzyme of LCFA oxidation.
- Inhibited by malonyl CoA ⇒ made by ACC2 in mitochondria of skeletal muscle, cardiac muscle, and liver with sole purpose to regulatE CPT I
- Fatty acyl-carnitine passes through the inner mitochondrial membrane via the carnitine acylcarnitine translocase.
- In the matrix, fatty acyl group transferred to mitochondrial CoA by CPT II.

Deficiencies in Carnitine System
- Rare deficiencies of CPT-I, CPT-II, and translocase
- Carnitine deficiency
- decreased synthesis (premature babies)
- defect in membrane transporter in muscle, heart, kidney
- Severity varies
- Treatment:
- Avoidance of fasting
- Diet low in LCFA
- dietary supplementation wtih MCTG and carnitine










