Ch 4 - Ch 6 Flashcards
4) The three-dimensional structure of proteins 5) protein function 6) enzymes
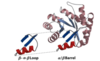
Peptide Bond
a peptide bond is a resonance hybrid of two canonical structures
- the peptide C–N bond is somewhat shorter than the C–N bond in a simple amine due to their partial double-bond character
the resonance causes the peptide bodes
- to be less reactive compared to esters
- to be quite rigid and nearly planar
- to exhibit a large dipole moment in the favored trans configuration
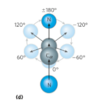
dihedral angles
the polypeptide is made up of a series of planes linked at alpha carbons
the dihedral angle is the angle at the intersection of two planes
peptide conformation is defined by three dihedral angles: phi, psi, and omega
some phi and psi combinations are very unfavorable because of steric crowding of backbone atoms with other atoms in the backbone or side chains
some phi and psi combinations are more favorable because of the chance to form favorable H-bonding interactions along the backbone
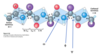
Ramachandran plot
shows the distribution of phi and psi dihedral angles that are found in a protein
shows the common secondary structure elements
reveals regions with the unusual backbone structure
dark blue areas represent conformations that involve no steric overlap and the white regions are conformations that are not allowed due to steric constraints

What are secondary structures?
secondary structure refers to any chosen segment of a polypeptide chain and describes the local spatial arrangement of the polypeptide backbone
common, regular arrangements: alpha-helix, beta-sheet, beta-turn
irregular arrangement: random coil
a-helix
- stabilized by hydrogen bonds between amides of an n and n+4 amino acids; h-bonds between the H on the nitrogen atom of a peptide linkage and the carbonyl oxygen atom of the fourth amino acid on the amino-terminal
- 3.6 residues per turn = 5.4 Å
- peptide bonds are aligned roughly parallel with the helical axis
- side chains point out and are roughly perpendicular with the helical axis
- right-handed alpha-helix is the common form
- large macroscopic dipole moment; negatively charged residues often occur near the positive end of the helix dipole

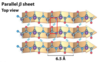
b-sheet
- sheet-like arrangement of backbone stabilized by hydrogen bonds between adjacent segments that may not be nearby
- side chains protrude from the sheet alternating in up and down direction
- parallel or antiparallel orientations are possible
Which amino acid residues are helix formers and which are helix breakers?
Ala and Leu are strong helix formers
Pro and Gly are helix breakers
with Pro, rotation around the N-Calpha bond is impossible
with Gly, the tiny R-group supports other conformations
attractive or repulsive interactions between side chains 3-4 amino acids apart will affect the formation (charge, bulk, and shape)
parallel b-sheets
the H-bonded strands run in the same direction (weaker H-bonds)
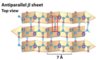
antiparallel b-sheets
the H-bonded strands run in opposite directions (stronger H-bonds)
ß turns
connect the ends of two adjacent segments of an antiparallel ß sheet
the 180º turn involves 4 amino acids; the turn is stabilized by a hydrogen bond from carbonyl oxygen to amide proton in the 4th residue
proline in position 2 (more common) or glycine in position 3 are common in ß turns
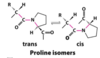
Pro: can be in the cis conformation
Gly: because R-group is small, so flexible
often found near the surface of a protein where the two central amino acid residues in the turn hydrogen-bond with water
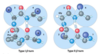
proline isomers
most peptide bonds not involving proline are in the trans configuration (> 99.95%)
about 6% of proline peptide bonds are in the cis configuration. most of the 6% involve ß turns
proline isomerization is catalyzed by proline isomerases
What is the length of a polypeptide with 80 amino acid residues in a single, continuous alpha-helix?
120 Å
an idealized alpha-helix has 3.6 residues per turn and the rise along the helical axis is 5.4 Å
Circular dichroism spectroscopy (CD)
a measurement of the difference in absorption of left-handed and right-handed circularly polarized light
The light-absorbing entity (chromophore) is the peptide bond; a signal is obtained when the peptide bond is in a folded environment
The alpha-helix and b-conformations have characteristic CD spectra (see figure)
a method for assessing common secondary structure
- can see if proteins are properly folded
- estimate the fraction of the protein that is folded in either of the common secondary structures
- monitor transitions between the folded and unfolded states

Tertiary Structure
- stabilized by numerous weak interactions between amino acid side chains
- largely hydrophobic and polar interactions
- can be stabilized by disulfide bonds
- two major classes: fibrous and globular (water- or lipid-soluble)
What are the different types of fibrous proteins?
a-Keratin (a-helix)
Silk (beta conformation)
collagen (triple helix)
all fibrous proteins have high concentration of hydrophobic amino acid residues both in the interior and on the surface of the protein
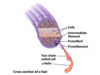
Keratin
right-handed a-helix crosslinked by disulfide bonds
two a-keratin strands oriented in parallel are wrapped around each other to form a left-handed coiled-coil
tough, insoluble protective structures of varying hardness and flexibility
found only in mammals (hair, wool, nails, claws, horns, hooves, and the outer layer of skin)
chemistry of perms: reduce cystine residues to break disulfide bonds, hair is manipulated (straight or curled), then oxidized to reform disulfide bridges
silk
fibroin is the main protein in silk from moths and spiders
antiparallel beta-sheet: small side chains (ala and gly) allow close packing of sheets that are stabilized by hydrogen bonding and van der Waal’s interactions
extremely strong (stronger than steel), soft, flexible filaments
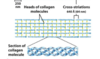
collagen
three pro- and gly-rich left-handed collagen helices intertwine into a right-handed superhelical triple helix
many triple helices assemble into a collagen fibril
in tendons, cartilage, cornea, and bone matrix
high tensile strength (higher than a steel wire of equal cross-section), without stretch
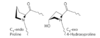
Scurvy and Vit C
scurvy is caused by a lack of Vitamin C (ascorbate)
Vitamin C is required for the hydroxylation of proline and lysine in collagen; hydroxylation forces the proline ring into a conformation that allows more hydrogen bonds between the three strands of collagen
without ascorbate, degeneration of connective tissue (hemorrhage, tooth loss, poor wound healing and reopening of old wounds, bone pain and degeneration, and eventually heart failure)
What kind of tertiary structures are in globular proteins?
- proteins are made of different motifs folded together
- motifs (folds): the specific arrangement of several secondary structures
- beta-a-beta loop
- alpha/beta-barrel
- can be found as reoccurring structures in numerous proteins
- domains are an assembly of motifs that can stand alone, often with a separable biological function
x-ray crystallography
Steps needed
- purify the protein
- crystallize the protein
- collect diffraction data
- calculate electron density; regions of greatest electron density reveal the location of atomic nuclei
- fit residues into density
Pros: no size limits; well-established
Cons: difficult for membrane proteins; cannot see hydrogens
The first globular protein structure to be determined by x-ray diffraction was that of myoglobin.
What are the three protein structure methods you need to know?
1) circular dichroism spectroscopy (CD)
2) x-ray diffraction
3) NMR
NMR
steps needed
- purify the protein
- dissolve the protein
- collect NMR data
- assign NMR signals
- calculate the structure: computer generates a family of closely related structures that represent the range of conformations consistent with the distance constraints
Pros: no need to crystallize the protein; can see many hydrogens
Cons: difficult for insoluble proteins; works best with small proteins
intrinsically disordered proteins
- contain segments that lack definable structure; lack hydrophobic core
- composed of high concentrations of Lys, Arg, Glu, and Pro, which force less-defined structure
- disordered regions can conform to many different proteins, facilitating interaction with numerous different partner proteins
- PONDER (predictor of natural disordered regions): the higher the PONDR score (0 - 1.0) the more intrinsic disorder
proteostasis
the continual maintenance of cellular protein activity accomplished by the coordination of many different pathways
3 kinds of processes contribute to proteostasis:
1) protein synthesis
2) protein folding
3) protein degradation
improperly folded or unfolded protein may aggregate and contribute to disease and aging processes

protein denaturation
denaturation: loss of structural integrity with accompanying loss of activity
proteins can be denatured by
- heat or cold
- pH extremes
- organic solvents
- chaotropic agents: urea and guanidinium hydrochloride
what did the ribonuclease refolding experiment prove?
the sequence alone determines the native conformation of proteins
Experiment
- ribonuclease is a small protein that contains 8 cysteines linked via four disulfide bonds
- ribonuclease denatures fully with urea and 2-mercaptoethanol
- when urea and 2-mercaptoethanol are removed, the protein spontaneously refolds and the correct disulfide bonds are reformed
Levinthal’s paradox
it is mathematically impossible for protein folding to occur by randomly trying every possible conformation until the lowest-energy one is found
How do proteins fold?
not random; follow a distinct path toward the native structure that’s thermodynamically most favorable
1) secondary structures form first (ionic interactions play an important role in guiding these early folding steps)
2) motifs are formed (hydrophobic effect plays a significant role throughout the process)
3) domains fold (domains near the amino terminus are synthesized first)
4) entire polypeptide is folded

The protein folding process viewed as a free-energy funnel

a) multiple folding pathways (folding order somewhat random); no stable folding intermediates; one stable native structure
b) represents a more typical protein; multiple possible stable folding intermediates on the multiple folding pathways; possibly several stable native structures
c) one stable native structure; no stable folding intermediates; few folding pathways
d) stable folding intermediates on every pathway leading to one native state (e.g. motif or domain always folds quickly, but other parts of the protein fold more slowly and in random order)
Chaperones
proteins that interact with partially folded or improperly folded polypeptides; facilitate correct folding pathways or provide microenvironments in which folding can occur
amyloid fibrils and human diseases
a soluble protein that is normally secreted from the cell is secreted in a misfolded state; most of these proteins have a concentration of aromatic amino acid residues in a core region of beta-sheet or a-helix
the core folds into a beta-sheet before the rest of the protein folds correctly, and the beta-sheets from two or more incompletely folded protein molecules associate to begin forming an amyloid fibril
other parts of the protein then fold differently, remaining on the outside of the b-sheet core in the growing fibril
the fibers are highly ordered and unbranched
amyloidoses: diseases like type 2 diabetes, Alzheimer, Huntington disease, and Parkinson disease associated with amyloid fibers

a) what is the molecular basis for the correlation between disulfide-bond content and mechanical properties of the protein?
b) most globular proteins are denatured and lose their activity when briefly heated to 65ºC. However, globular proteins that contain multiple disulfide bonds often must be heated longer at higher temperatures to denature them. On cooling a solution of these proteins, the activity of the protein is restored. What is the molecular basis for this property?
a) disulfide bonds are covalent bonds, which are much stronger than the noncovalent interactions that stabilize most proteins. They cross-link protein chains, increasing their stiffness, mechanical strength, and hardness.
b) Cystine residues (disulfide bonds) prevent the complete unfolding of the protein
A series of torsion angles, ϕ and ψ, that might be taken up by the peptide backbone is shown below. Which of these closely correspond to ϕ and ψ for an idealized collagen triple helix? (collagen triple helix is in quadrant II (-45,160) of Ramachandran plot)

φ = (f) and Ψ = (e)
a) where might bends or ß turns occur?
b) where might intrachain disulfide cross-linkages be formed?
c) assuming that this sequence is part of a larger globular protein, indicate the probable location (external surface or interior of the protein) of the following amino acid residues: Asp, Ile, Thr, Ala, Gln, Lys. Explain your reasoning.

a) bends are most likely at 7 and 19; pro residues in the cis configuration accommodate turns well
b) cys residues at 13 and 24 can form disulfide bonds
c) external surface: polar and charged residues (Asp, Gln, Lys)
interior: nonpolar and aliphatic (Ala, Ile)
Thr has a hydropathy index near zero thus can be found either on the external surface or in the interior of the protein
A sample of (660 mg) of an oligomeric protein of Mr 132,000 was treated with an excess of Sanger’s reagent under slightly alkaline conditions until the chemical reaction was complete. The peptide bonds of the protein were then completely hydrolyzed by heating it with concentrated HCl. The hydrolysate was found to contain 5.5 mg of the following compound (see figure).
2,4-dinitrophenyl derivatives of the a-amino groups of other amino acids could not be found.
a) Explain how this information can be used to determine the number of polypeptide chains in an oligomeric protein.
b) Calculate the number of polypeptide chains in this protein.
c) What other analytic technique could you employ to determine whether the polypeptide chains in this protein are similar or different?

a) Because only a single 2,4-dinitrophenyl (DNP) amino acid derivative is found, there is only one kind of amino acid at the amino terminus (i.e. all the polypeptide chains have the same amino-terminal residue)
comparing the number of moles of DNP-valine to the number of moles of protein equals the number of amino termini and thus the number of polypeptide chains
b) 4
c) different chains would probably run as discrete bands on an SDS PAGE.
Protein A has a binding site for ligand X with a Kd of 10–6 M. Protein B has a binding site for ligand X with a Kd of 10–9 M. Which protein has a higher affinity for ligand X? Explain your reasoning. Covert the Kd to Ka for both proteins.
Protein B has a higher affinity for ligand X; it will be half-saturated at a much lower concentration of X than will protein A.
Protein A has Ka = 106 M–1
Protein B has Ka = 109 M–1
(when [L] = Kd, half of the ligand-binding sites are occupied)
What is the effect of the following changes on the O2 affinity of hemoglobin?
a) a drop in the pH of blood plasma from 7.4 to 7.2.
b) a decrease in the partial pressure of CO2 in the lungs from 6 kPa (holding one’s breath) to 2 kPa (normal breathing).
c) an increase in the BPG level from 5mM (normal altitudes) to 8 mM (high altitudes).
d) an increase in CO from 1.0 part per million (ppm) in a normal indoor atmosphere to 30 ppm in a home that has a malfunctioning or leaking furnace.
a) decreases
b) increases
c) decreases
d) decreases
The affinity of Hb for O2 is regulated by the binding of the ligands H+, CO2, and BPG. The binding of each ligand shifts the O2-saturation curve to the right – that is, the O2 affinity of hemoglobin is reduced in the presence of ligand.
a binding protein binds to a ligand L with a Kd of 400 nM. How much ligand is present when Θ is
a) 0.25
b) 0.6
c) 0.95
Θ = the fraction of occupied binding sites
a) 0.13 pM
b) 0.6 pM
c) 7.6 µM
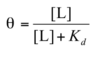
Under appropriate conditions, hemoglobin dissociates into its four subunits. The isolated alpha subunit binds oxygen, but the O2-saturation curve is hyperbolic rather than sigmoid. In addition, the binding of oxygen to the isolated alpha subunit is not affected by the presence of H+, CO2, or BPG.
What do these observations indicate about the source of the cooperativity in hemoglobin?
The observations indicate that the cooperative behavior of hemoglobin arises from the interaction between subunits.
O2 cooperative binding is graphically represented as a sigmoid curve. Noncooperative binding like that of myoglobin is hyperbolic.
BPG bind in the cavity between the ß subunits in the T state.
The four polypeptide chains of Hb communicate with each other about H+ binding to His of the ß subunits. When protonated, His forms an ion pair to Asp that helps stabilize the T state (deoxyhemoglobin).
CO2 binds as a carbamate group to the Hb and releases a H+, contributing to the Bohr effect. The bound carbamate also forms additional salt bridges that help to stabilize the T state and promote the release of oxygen.
A team of biochemists uses genetic engineering to modify the interface region between hemoglobin subunits. The resulting hemoglobin variants exist in the solution primarily as alpha-beta dimers (few, if any, a2ß2 tetramers form).
Are these variants likely to bind oxygen more weakly or more tightly? Explain your answer.
More tightly.
An inability to form tetramers would limit the cooperativity of these variants, and the binding curve would become more hyperbolic. Also, the BPG-binding site would be disrupted. BPG binds in the cleft between the two ß subunits. Oxygen binding would probably be tighter because the default state in the absence of bound BPG is the tight-binding R state.
Bohr effect
The effect of pH and CO2 concentration on the binding and release of oxygen by hemoglobin.
The binding of H+ (amino acid residues, particularly His146 in beta subunits) and CO2 (amino-terminal end of each globin chain) is inversely related to the binding of oxygen. At the relatively low pH and high CO2 concentration of peripheral tissues, the affinity of hemoglobin for oxygen decreases as H+ and CO2 are bound, and O2 is released to the tissues.
Hemoglobin Cooperative Binding
O2 binding to one subunit affects the affinity for O2 in adjacent subunits.
the first molecule of O2 that interacts with deoxyhemoglobin binds weakly because it binds to a subunit in the T state.
Its binding leads to conformational changes that are communicated to adjacent subunits, making it easier for additional O2 molecules to bind. T→R transition occurs more readily in the second subunit.
The last O2 molecule binds to a heme in a subunit that is already in the R state, and thus binds with much higher affinity than the first molecule.
Two proteins, A and B, bind to the same ligand, L, with the binding curves shown below.
What is the dissociation constant, Kd, for each protein?
Which protein (A or B) has a greater affinity for ligand L?

The concentration of ligand at which half the binding sites are occupied is the dissociation constant.
A, Kd = 2 µM
B, Kd = 6 µM
Because A is half-saturated at a lower [L], it has a higher affinity for the ligand.
Hill Plot
Quantitative Description of Cooperativity
n = Hill coefficient (the degree of cooperativity)
n = 1 → no cooperativity (myoglobin)
n > 1 → positive cooperativity (hemoglobin)
n < 1 → negative cooperativity

The researchers working on happyase discover that the compound STRESS is a potent competitive inhibitor of happyase.
Addition of 1 nM STRESS increases the measured Km for SAD by a factor of 2. What are the values of a and a’ under these conditions?
The Km measured in the presence of a competitive inhibitor is defined as aKm.
By definition, the value of a’ for a competitive inhibitor is 1.
a = 2; a’ = 1
Strong Binding and Weak Binding
Kd values?
Strong binding: Kd < 10 nM
weak binding: Kd > 10 µM
Lock and Key binding
complementary surfaces of the binding site and ligand are preformed
What determines the high specificity of proteins?
Complementary of the binding sites and the ligand.
complementary in
size
shape
charge
or hydrophobic/hydrophilic character
Induced Fit Binding
conformational changes occur upon ligand binding; both the ligand and the protein can change their conformations
allows for tighter binding of the ligand
allows for high affinity for different ligands
Myoglobin: CO binding and structure
CO has a similar size and shape to O2 and fits the same binding site
CO binds over 20,000 times better than O2 because the carbon in CO has a filled lone electron pair that can be donated to vacant d-orbitals on the Fe2+
Protein pocket decreases affinity for CO, but it sill binds about 250 times better than oxygen; histidine residue in pocket hydrogen bonds with O2
CO is highly toxic as it competes with oxygen and blocks the function of myoglobin, hemoglobin, and mitochondrial cytochromes that are involved in oxidative phosphorylation

Why do we need oxygen-binding proteins?
Protein side chains lack affinity for O2
Some transition metals bind O2 well but would generate free radicals if free in solution
organometallic compounds such as heme are more suitable, but Fe2+ in free heme could be oxidized to irreversible Fe3+
Capture oxygen with heme that is protein-bound
Conformational change from the T state to the R state involves what?
Breaking ion pairs between the a1 - b2 interface
pH effect on O2 binding to hemoglobin
actively metabolizing tissues generate H+, lowering the pH of the blood near the tissues relative to the lungs
- CO2 produced by metabolism is exported in the form of a carbamate, which yields a proton
- the rest of the CO2 is exported as dissolved bicarbonate, also producing a proton
H+ bind to Hb and stabilizes the T state, releasing O2 in the tissues
- carbamate forms additional salt bridges, stabilizing the T-state
2,3-bisphosphoglycerate (BPG)
negative heterotropic regulator of Hb
present at mM concentrations in erythrocytes (produced from an intermediate in glycolysis)
small negatively charged molecule binds to the positively charged central cavity of Hb, stabilizing the T states
allows for O2 release in the tissues and adaptation to changes in altitude; maintains the same relative difference in O2 bound and unbound Hb
Immunoglobulin G
antibody composed of 2 heavy chains and 2 light chains
light chains: one constant and one variable domain
heavy chains: three constant and one variable domain
variable domains make up antigen-binding site (two per antibody) that are hypervariable (confers high antigen specificity)
antigens bind via induced fit
sickle-cell anemia
Glu6 → Val in the beta-chain of Hb
the new Val side chain can bind to a different Hb molecule to form a strand, sickling the red blood cells
untreated homozygous individuals generally die in childhood
heterozygous individuals exhibit a resistance to malaria
the specificity of the antibody has important analytical use
name the two tools and describe how they work
column chromatography and ELISA

protein interactions modulated by chemical energy (ATP)
can cause conformational changes in proteins, generally required for their function (e.g. for motor proteins that control the movement of cells and organelles)
allows for spatial and temporal regulation of interactions
muscle structure
muscle fiber: large, single, elongated multinuclear cell
each fiber contains about 1,000 myofibrils
myofibrils contain thick filaments (myosin) and thin filaments (actin); divided into sarcomeres (contractile unit)
I band contain only thin filaments; A band is where parallel thick and think filaments overlap. myosin thick filaments slide along actin thin filaments
Z disk serves as an anchor to which the thin filaments are attached; M line is a region of high electron density in the middle of the thick filaments

actomyosin cycle
muscle contraction caused by a series of conformational changes to protein structure

regulation of muscle contraction
availability of myosin-binding sites on actin is regulated by troponin and tropomyosin (avoids continuous muscle contraction)
nerve impulse triggers the release of Ca2+; causes conformational changes to tropomyosin-troponin complex, exposing myosin-binding sites
What are enzymes? What do they do/not do?
Enzymes are catalysts
They increase reaction rates without being used up by lowering the activation barrier (the free energy of the transition state)
they do not affect equilibrium (∆G); any enzyme that catalyzes the reaction S → P also catalyzes the reaction P→S. The reaction reaches equilibrium much faster because the rate of reaction is increased.

Most enzymes are which macromolecule? Others are?
Most enzymes are globular proteins
Some RNA (ribozymes and ribosomal RNA) also catalyze reactions
Why biocatalysis over inorganic catalysts?
- Greater reaction specificity: avoids side products; metabolites have many potential pathways of decomposition; enzymes make the desired one most favorable
- milder reaction conditions: conducive to the cell environment
- higher reaction rates: in a biologically useful timeframe
- capacity for regulation: control of biological pathways
six classes of enzymes
defined by the reactions catalyzed
- oxidoreductases: transfer of electrons (H atoms)
- transferases: group transfer reactions
- hydrolases: hydrolysis reactions (transfer of functional groups to water)
- lyases: cleavage of C-C, C-O, C-N by elimination leaving double bonds or addition of groups to double bonds
- isomerases: transfer of groups within molecules to give isomeric forms
- ligases: formation of C-C, C-S, C-O, and C-N bonds by condensation reactions coupled with expenditure of energy from ATP or other similar cofactor
Explain this diagram

The equilibrium between S and P reflects the difference in the free energies of their ground states.
The free energy of the ground state of P is lower than that of S, so ∆G° for the reaction is negative (the reaction is exergonic) and at the equilibrium, there is more P than S. The position and direction of equilibrium are not affected by any catalyst.
The transition state indicates a point at which decay to the S or P state is equally probable (it is downhill either way).
The difference between the energy levels of the ground state and the transition state is the activation energy, ΔG‡. Higher activation energy corresponds to a slower reaction.

Is the transition state a rection intermediate?
No
The transition state is not a chemical species with any significant stability and should not be confused with a reaction intermediate (such as ES or EP).
A transition state is simply a fleeting molecular moment (that happens after ES forms) in which events such as bond breakage, bond formation, and charge development have proceeded to the precise point at which decay to substrate or decay to product are equally likely.
Rate-limiting step?
when several steps occur in a reaction, the overall rate is determined by the step (or steps) with the highest activation energy
for enzymes with several steps that have similar activation energies, all these steps would be partially rate-limiting
what is the purpose of an activation barrier?
without energy barriers, complex macromolecules would revert spontaneously to much simpler molecular forms, and the complex and highly ordered structures and metabolic processes of cells could not exist
How do enzymes lower the activation energy?
- uncatalyzed bimolecular reaction
- converting two reactants into a single restricted transition state is entropically unfavorable
- uncatalyzed unimolecular reactions
- combining two ends of a molecule that’s super flexible into a rigid transition state is also entropically unfavorable
- Catalyzed reactions
- binding energy is a major source of free energy used by enzymes to lower the activation energies of reactions
- binding energy (∆GB) comes from the formation of the ES complex by forming weak, noncovalent interactions (H-bonds, ionic interactions, and hydrophobic effect)
- each weak interaction is accompanied by the release of a small amount of free energy that stabilizes the interaction
- entropy cost (decreased entropy) from organizing reactive groups into close proximity and proper orientation is paid by the binding energy
- weak intereactions between enzyme and susbstrate are optimized in transition state
What are the different catalytic mechanisms and how are they different from electrostatic catalysis?
acid-base catalysis: transfer of protons
covalent catalysis: change reaction pathway
metal ion catalysis: use redox cofactors, pKa shifters
electrostatic catalysis: preferential interactions with TS
the electrostatic mechanism involves binding energy, whereas the others involve transient covalent interaction with a substrate or group transfer to or from a substrate
Optimal interactions between substrate and enzyme occur at which state?
Explain the TS Stabilization Idea with the Imaginary Stickase
transition state
enzyme active sites are complementary to the transition state of the reaction; therefore, bind better to TS than substrates
Sticakse
- before we get to the product (broken sticks), the substrate (metal stick) needs to be bent (transition state)
- lock-and-key model: stickase with magnet-lined pocket complementary in structure to the substrate stabilizes the substrate; the result is an increase in activation energy (∆GM)
- induced-fit model: an enzyme with a pocket complementary to the reaction TS helps destabilize the stick, contributing to catalysis of reaction. The binding energy of the magnetic interactions compensates for the increase in free energy required to bend the stick (lowering the activation energy - ∆GM)

general acid-base catalysis
amino acid side chains (titratable groups) take on the role of proton donors and acceptors
also can be prosthetic groups and cofactors

covalent catalysis
a transient covalent bond between the enzyme and the substrate
changes the reaction pathway
requires a nucleophile on the enzyme: serine, thiolate, amine, or carboxylate
metal ion catalysis
involves a metal ion bound to the enzyme
interacts with substrate to facilitate binding; stabilizes negative charges
participates in oxidation reactions
What is enzyme kinetics
determines the rate of a reaction and how it changes in responses to changes in experimental parameters: enzyme, substrate, effectors, and temperature
we typically look at the appearance of the product
How do you do a kinetic experiment?
1) test tube: mix enzyme + substrate
2) start taking samples where we measure the absorbance of the product (the appearance of the product), using a spectrophotometer, to get a continuous line
3) measuring the slope (initial velocity) of the beginning of the reaction (where substrate concentration changes very little). This is where we can get a linear response to the substrate concentration. The slope shows us how fast the enzyme is working at that particular [s]; with increasing substrate concentration, the slope (initial velocity) increases
4) change substrate concentration and repeat to get different initial rates
5) once you have a whole bunch of initial rates, you plot them on the Michaelis-Menten plot, which shows us how the initial rate changes with an increasing substrate concentration; also allows us to determine Vmax and Km

why study enzyme kinetics?
you do a single experiment and get a ton of information out of it:
a quantitative description of biocatalysis
determine the order of binding of substrates
elucidate acid-base catalysis
understand catalytic mechanism
find effective inhibitors
understand regulation of activity
Effect of Substrate Concentration
at relatively low [S], initial velocity increases almost linearly with an increase in [S] (the more substrate, the faster the reaction goes)
at higher substrate concentrations, initial velocity increases by smaller and smaller amounts in response to increases in [S] eventually reaching a plateau-like V0 region (changes in V0 are super small as [S] increases)
V0 approaches but never quite reaches maximum velocity (Vmax)
the substrate concentration at which V0 is half-maximal is Km (Michaelis constant)

What can go wrong with the kinetics experiment?
substrate prep contains inhibitors
enzyme prep contains inhibitors
substrate inhibition
limitation of measurements
saturation/steady-state kinetics
The Vmax for a catalyzed reaction is observed when the enzyme is “saturated” with its substrate. Increases in [S] has no effect on the initial rate. Essentially all free enzymes have been converted to the ES form. After one enzymatic turnover, P is generated at the same rate that S is consumed; [ES] remains constant.
The measured V0 generally reflects the steady-state and analysis of these initial rates is referred to as steady-state kinetics.
Michaelis-Menten equation
the rate-limiting step is the breakdown of the ES complex to product and free enzyme (k2); highly simplified assumption (one reactant, one product, no inhibitors)
- at low [S], Km>>[S], and the [S] term in the denominator of the Michaelis-Menten equation becomes insignificant; V0 exhibits a linear dependence on [S]
- at high [S], where [S] >> Km, the Km term in the denominator becomes insignificant and the equation simplifies to V0=Vmax

Lineweaver-Burke Plot
linearized double-reciprocal plot
good for analysis of two-substrate data or inhibition
also allows a more accurate determination of Vmax, which can only be approximated by the Michaelis-Menten plot

when Km=Kd
Km can vary greatly from enzyme to enzyme, and even for different substrates of the same enzyme
Km only represents the binding affinity (dissociation constant Kd) when the rate-limiting step is k2 (ES → E + P)
more common are cases in which the reaction goes through several steps after formation of ES; Km then becomes a very complex function of many rate constants
Vmax reflects the rate-limiting step in a reaction
kcat
turnover number (unitless)
how many substrate molecules can one enzyme molecule convert per second; equivalent to the rate constant for the rate-limiting step
kcat = Vmax / [Etot]

specificity constant and upper limit
the best way to compare the catalytic efficiencies of different enzymes or the turnover of different substrates by the same enzymes
high velocity vs high affinity; you can’t have both
kcat / Km
diffusion-controlled limit (ideal efficiency): the rate at which E and S can diffuse together in an aqueous solution (108 to 109 M-1s-1); many enzymes have a specificity constant near this range; such enzymes are considered to be perfect
A team of motivated researchers sets out to study the enzyme, which they call happyase. They find that the kcat for happyase is 600 s-1. When [Et] = 200 nM and [SAD] = 40 µM, the reaction velocity, is 9.6 µM s-1.
Calculate Km for the substrate SAD.

Km = 10 µM

A team of motivated researchers sets out to study the enzyme, which they call happyase. they find the kcat for happyase is 600 s-1.
[Et] = 10 mM, V0 = 3 µM s-1, Km = 10 µM
What is the [S] used in this experiment?

[S] = 10 µM
two-substrate reactions
most enzyme reactions are not simple reactions; they involve 2 substrates
- sequential reaction: form a ternary complex
- random order: both active sites already open
- ordered: S1 binds before S2 (conformational change for binding site 2)
- ping-pong reaction: no ternary complex is formed
- the first substrate is converted to product and dissociates before the second substrate binds
- part of the S1 gets left behind in the active site; the altered enzyme interacts with S2
- ex: transaminase moves an amino acid to E to form a covalently modified enzyme intermediate and pyruvate (p1) is released; alphaketoglutarate (s2) binds to modified enzyme; amine left behind is transferred to s2, which results in free enzyme and glutamic acid

can we distinguish sequential reaction from ping-pong reaction?
yes with Lineweaver-Burke plot
the concentration of substrate 1 is varied while the concentration of substrate 2 is held constant. This is repeated for several values of [S2], generating separate lines
sequential reaction: lines intersect
Ping pong reaction: parallel lines

What are enzyme inhibitors and what are the two types?
inhibitors are compounds that decrease the enzyme’s activity
- irreversible inhibitors (inactivators)
- bind covalently with or destroy a functional group on an enzyme that’s essential for enzyme activity
- form a highly stable noncovalent association
- often powerful toxins; also used as drugs
- reversible inhibitors bind to and can dissociate from the enzyme
- often structural analogs of substrates or products
- used as drugs to slow down a specific enzyme
- competitive, uncompetitive, and mixed inhibitors
competitive inhibition
resembles the substrate and both bind to the active site (increase in Km)
does not affect catalysis (no change in Vmax)
Lineweaver-Burke: lines produced by different inhibitor concentrations intersect at the y-axis

uncompetitive inhibition
- only binds to ES complex; does not affect substrate binding
- inhibits catalytic function; a decrease in Vmax
- an apparent decrease in Km; because the [S] required to reach one-half Vmax decreases by the factor a’
- no change in Km/Vmax
- Lineweaver-Burke: lines are parallel

mixed inhibition
- binds enzyme at a site distinct from the active site, but binds to either E or ES
- inhibits catalysis by lowering the [E] (decrease in Vmax)
- the Km may increase or decrease depending on which enzyme form E or ES the inhibitor binds to most strongly
- Lineweaver-Burke: lines intersect left of the y-axis
- noncompetitive inhibitors are mixed inhibitors such that there is no change in Km; these are rare

transition state analogs
stable molecules that are designed to resemble reaction transition states
bind to an enzyme more tightly than does the substrate in the ES complex because they fit into the active site better (form a greater number of weak interactions)
allosteric regulation
in many allosteric enzymes, the substrate-binding site and the modulator-binding sties are on different subunits (catalytic and regulatory subunits)
homotropic regulation: hemoglobin
allosteric modulators should not be confused with uncompetitive and mixed inhibitors; although inhibitors bind at a second site on the enzyme, they do not necessarily mediate conformational changes between active and inactive forms
usually found at the top of the metabolic pathway and at branch points

zymogen activation
zymogen: inactive precursor; cleaved to form the active enzyme (via conformational change)
many proteases of the stomach and pancreas are regulated this way
active trypsin in the pancreas results in pancreatitis and possible pancreas cancer; trypsinogen is secreted into the lumen of our intestine and cleaved (proteolytic modification); trypsin modifies chymotrypsinogen to active chymotrypsin
this type of activation is irreversible; inactivated by inhibitor proteins that bind very tightly to the enzyme active site

enzyme activity can be regulated by…
noncovalent modification (allosteric effectors)
covalent modification (phosphorylation, ubiquitination)
irreversible
reversible
gene expression (making more/fewer enzymes)
protein degradation
aspartate transcarbamoylase (ATCase)
catalyzes an early step in the biosynthesis of pyrimidines
highly regulated by the end-products of pyrimidine synthesis (CTP); binding of CTP results in T state of the enzyme (feedback inhibition); at low levels of CTP, CTP comes off the enzyme, and the enzyme undergoes a T to R transition

The kinetics of allosteric regulators: hemoglobin vs ATCase
myoglobin: high-activity enzyme
hemoglobin: high activity state to low-activity state (R to T transition from lungs to tissues)
ATCase: from CTP bound (low affinity), as we have a decreasing amount of CTP in the cell, the population of active ATCase increases (more and more high-affinity R state)

The enzyme urease enhances the rate of urea hydrolysis at pH 8.0 and 20°C by a factor of 1014. If a given quantity of urease can completely hydrolyze a given quantity of urea in 5.0 min at 20°C and pH 8.0, how long would it take for this amount of urea to be hydrolyzed under the same conditions in the absence of urease?
assume that both reactions take place in sterile systems so that bacteria cannot attack the urea.
9.5 x 108 years
When enzyme solutions are heated, there is a progressive loss of catalytic activity over time due to the denaturation of the enzyme. A solution of the enzyme hexokinase incubated at 45°C lost 50% of its activity in 12 min, but when incubated at 45°C in the presence of a very large concentration of one of its substrates, it lost only 3% of its activity in 12 min.
Suggest why the thermal denaturation of hexokinase was retarded in the presence of one of its substrates.
The ES complex is more stable than the free enzyme. The ground state for the ES complex is at a lower energy level than that for the free enzyme, thus increasing the height of the energy barrier to be crossed to get from the native to the denatured state.
Which of the listed effects would be brought about by an enzyme catalyzing the following simple reaction?
a) decreased K’eq
b) increased k1
c) increased K’eq
d) increased ΔG‡
e) decreased ΔG‡
f) more negative ΔG°
g) increased k2

b, e, g
a) at what substrate concentration would an enzyme with a kcat of 30.0 s-1 and a Km of 0.0050 M operate at one-quarter of its maximum rate?
b) Determine the fraction of Vmax that would be obtained at the following substrate concentrations: 1/2Km, 2Km, and 10Km.
c) An enzyme that catalyzes the reaction X ⇌ Y is isolated from two bacterial species. The enzymes have the same Vmax but different Km values for the substrate X. Enzyme A has a Km of 2.0 µM and enzyme B has a Km of 0.5 µM. The plot below shows the kinetics of reactions carried out with the same concentration of each enzyme and with [X] = 1 µM. Which curve corresponds to which enzyme?

a) 1.7 x 10-3 M
b) 0.33; 0.67; 0.91
c) The upper curve = enzyme B; the lower curve = enzyme A.
When the initial concentration of the substrate is greater than Km, the rate of the reaction is less sensitive to the depletion of the substrate at the early stages of the reaction and the rate remains approximately linear for a longer time.
An enzyme catalyzes the reaction A ⇌ B. The enzyme is present at a concentration of 2 nM, and the Vmax is 1.2 μM s−1. The Km for substrate A is 10 μM.
Calculate the initial velocity of the reaction when the substrate concentration is
(a) 2 μM, (b) 10 μM, (c) 30 μM
a) 0.2 μM s−1
b) 0.6 μM s−1
c) 0.9 μM s−1
Prostaglandins are derived from the 20-carbon fatty acid arachidonic acid in a reaction catalyzed by the enzyme prostaglandin-endoperoxide synthase. This enzyme, a cyclooxygenase, uses oxygen to convert arachidonic acid to PGG2, the immediate precursor of many different prostaglandins.
(a) The kinetic data given below are for the reaction catalyzed by prostaglandin-endoperoxide synthase. Focusing here on the first two columns, determine the Vmax and Km of the enzyme.
(b) Ibuprofen is an inhibitor of prostaglandin-endoperoxide synthase. By inhibiting the synthesis of prostaglandins, ibuprofen reduces inflammation and pain. Using the data in the first and third columns of the table, determine the type of inhibition that ibuprofen exerts on prostaglandin-endoperoxide synthase.

a) Calculate the reciprocal values and draw a double-reciprocal plot to determine the kinetic parameters
Vmax = 51.5 mM/min
Km = 0.59 mM
b) competitive inhibitor; in the Lineweaver-Burke plot, the Vmax is unchanged and Km is increased

The active site of lysozyme contains two amino acid residues essential for catalysis: Glu35 and Asp52. The pKa values of the carboxyl side chains of these residues are 5.9 and 4.5, respectively.
What is the ionization state (protonated or deprotonated) of each residue at pH 5.2, the pH optimum of lysozyme?
How can the ionization states of these residues explain the pH-activity profile of the lysozyme shown below?
at pH 5.2, Asp52 is mainly deprotonated, whereas Glu35 is protonated.
The pH activity profile suggests that maximum catalytic activity occurs at a pH midway between the pKa values of the two acidic groups when Glu35 is protonated and Asp52 is deprotonated.

