3- Introduction to White Blood Cell Disorders, Reactive and Neoplastic Myeloid Processes Flashcards
WHat WBCs are in the bone marrow?
Pluripotent stem cells
Lymphoblast
Myeloid blast Erythroid precursor
Megakaryocyte
Myeloblast

What WBCs are in the peripheral blood?
Monocyte
Neutrophil
Eosinophil
Basophil
Mature Lymphocyte

Where are WBCs distributed?
- bone marrow
- peripheral blood: granulocytes, monocytes, lymphocytes (B, T, NK)
- Lymph nodes, thymus, spleen, tonsils, adenoids, peyer patches
- Mucosa-associated lymphoid tissue (MALT): Lung, GI tract
What are benign leukocyte disorders? And how do we classify them?
Definition: NOT neoplastic (clonal)
- Classified as:
- Qualitative (structural/functional) disorders
- Quantitative (numerical) disorders
What are the 2 different types of quantitative (numerical) disorders?
Increased-cytoses
Decreased-cytopenias
What is a leukemoid reaction?
NOT leukemia! (benign) exaggerated response to infection
Absolute leukocyte count >50,000/uL
May involve neutrophils, lymphocytes or eosinophils
What are 3 common etiologies of leukemoid reactions?
Perforating appendicitis: Neutrophils
Whooping cough (Bordatella pertussis): Lymphocytes
Cutaneous larva migrans (nematodes): Eosinophils
What is a leukoerythroblastic reaction? What 2 things can it be due to?
Immature bone marrow cells in the peripheral blood (PB)
- Due to: BM infiltrative disease
- infiltrative disease: Fibrosis or metastatic breast cancer
- Due to severe BM stress
- sepsis or growth factor

What is neutrophilia? Wat are 3 possible etiologies? What are 2 mechanisms?
Absolute neutrophil count >7,000/uL
- Etiology
- infection (acute appendicitis)
- Sterile inflammation with necrosis (acute MI)
- Drugs (steroids, catecholamines, lithium)
- Mechanism
- increased production
- decreased margination

What is neutropenia?
Absolute neutrophil count <1,500/uL
- Etiology
- chemotherapy
- aplaastic anemia
- immune destruction (SLE)
- septic shock
- Mechanism
- decreased prodtuction increased estruction or margination
What is eosinophilia? What are 4 possible etiologies? What is the mechanism?
Absolut eeosinophil count > 700/uL
- Etiology
- Type I hypersensitivity (bronchial asthma, penicilin allergy, hay fever)
- Invasive helminths (stongyloidiasis, hook worm)
- Hypocortisolism (Addison’s disease)
- Neoplasm (Hodgkin lymphoma)
- Mechanism
- increased production (induced by interleukins)
- increased tissue recruitment by chemotactic factors

What is basophilia?
- Absolutel Basophil >200/uL
- Etiology
- Chronic myeloid leukemia (and other myeloproliferative neoplasms MPN)
- Hypersensitivity/inflammatory reactions
- Hypothyroidism
- Infections (TB, certain viruses)
- Chronic Kidney disease
What are the 2 Neoplastic leukocyte disorders?
Leukemia: Proliferation of neoplastic cells, primarily in BM and PB
Lymphoma: Proliferation of neoplastic cells, primarily in LNs and extramedullary lymphoid tisue
What are myeloid neoplasms? What are teh WHO classification criteria?
Neoplastic stem cell disorders, may involve one or more cell lineages
WHO: Morphology, Immunophenotype, Genetic features, Clinical features
What are 4 Myeloproliferative neoplasms?
Chronic Myeloid Leukemia, BCR-ABL1 posiitve (CML)
Polycythemia vera (PV)
Primary Myelofibrosis (PMF)
Essential thrombocytopenia (ET)
What are general features of MPN?
- Clonal hematopoietic stem cell disorders
- Proliferation of one or more of the myeloid lineages
- Granulocytic
- Erythroid
- Megakaryocytic
At what age do people usually get MPNs? Describe the cellularity of the bone marrow? Is it effective or ineffective hematopoiesis? What are 2 physical results? What are we worried about?
- Common in adults (5th-7th decade)
- Hypercellular BM with effective hematopoiesis (increased PB granulocytes, RBCs and/or platelets)
- You see splenomegaly or hepatomegaly
- Potential for progression (BM fibrosis or acute leukemia)
What is the difference between MPN and MDS?
- MPN: Hypercellular BM with effective hematopoiesis
- increased PB counts- cytoses
- clonal abnormalities increase cell proliferation
- MDS: Hypercellular BM with ineffective hematopoiesis
- decreased PB counts: cytopenias
- clonal abnormalities promoote cell death
What is the epidemiology of Chronic myelogenous leukemia (CML)?
What is the pathogenesis? What are the clinical findings?
- Epidemiology: peak at 40-60 years
- Pathogenesis:
- Neoplastic expansion of the pluripotential stem cell
- BCR-ABL1 fusion gene (produces protein with tyrosine kinase activity)
- Clinical findings:
- Hepatosplenomegaly
- fatigue, wekaness, weight loss, anorexia
What are the laboratory findings for CML?
Leukocytosis with immature myeloid cells
Few myeloblasts (2-3%)
Basopilia
Thrombocytosis (40-50% cases) or thrombocytopenia
Hypercellular BM (~100%) with granulocytic hyperplasia
Philadelphia chromosome: t(9;22)
BCR-ABL1 fusion gene (FISH or RT-PCR)
What is the equation for what BM cellularity should be?
100-age
What is the clinical course of CML?
3 stages
Chronic phase (~3 years)
Accelerated phase (~1 year)
Blast phase= acute leukemia (myeloid or lymphoblastic)
What are the therapies for CML?
- Allogenic stem cell transplant
-
BCR-ABL tyrosine kinase inhibitors
- Gleevec (imatinib mesylate)
- Dasatinib
- Nilotinib
What is polycythemia vera (PV)? What mutation is associated?
Neoplastic explanation of the pluripotential stem cell
increase in RBCs granulocytes and platelets
Janus 2 Kinase gene (JAK2) mutation in virtually all cases
What are the 4 clinical findings of PV?
- Splenomegaly
- thrombotic events due to hyper-viscosity (eg hepatic vein thrombosis)
- gout (increased uric acid due to increased cell breakdown)
- signs of increased hiatamine (released from mast cells in the skin)
- ruddy face
- pruritis after bathing
- peptic ulcer disease
What are the 6 laboratory findings associated with PV?
Increased RBC mass
leukocytosis
thrombocytosis
decreased erythropoietin (EPO)
Normal oxygen saturation (SaO2)
hypercellular BM with fibrosis in later stages
What are the major and minor diagnostic criteria?
- Major
- hemoglobin > 16.5 g/dL in men, 16.0 g/dL in women or other evidence of increased RBC volume
- BM biopsy showing hypercellularity for age with trilineage growth (panmyelosis) with prominent erythroid, granulocytic and myeloid proliferation with pleomorphic mature megakaryocytes
- presence of Jak2 mutation
- Minor criterion
- serum EPO level below the normal reference range
What is the clinical course of PV like?
- Conservative treatment (eg phlemobotomy) median survival of > 10 yrs
- most pts die fo thrombosis or hemorrhage
- 15-20% of pts. evolve to “spent phase” which is similar to primary myelofibrosis
- 2-3% of pts develop MDS or AML
- (without cytotoxic therapy; >10% if history of cytotoxic therapy)
What therapies are used for PV?
Phlebotomy
Low dose aspirin
Cytoreductive therapy: i.e. hydroxyurea, in high risk patients
What is primary myelofibrosis (PMF)
- Rapid development of BM fibrosis and extramedullary hematopoiesis (EMH) in the spleen, liver and lymph nodes
- Clinical findings
- fatigue, splenomegaly, hepatomegaly, fever, bone pain, night sweats

What are the laboratory findings in PMF?
- JAK2 mutation in 50-60% of cases (also CALR, MPL)
- BM fibrosis and clusters of atypical megakaryocytes
- peripheral blood leukocytosis
- thrombocytosis (early)
- normochromic, mormocytic anemia
- teardrop cells
- leukoerythroblastic reaction

What is the clinical course of PMF?
- Median survival
- How many patients develop AML
- Major causes of morbidity and mortality
- Survival
- 3-7 yrs in fibrotic stage
- >10 yrs in early (prefibrotic) stage
- 5-30% of patients develop AML
- Major causes of morbidity and mortality
- BM failure (infection, hemorrhage)
- thromboembolic events
- portal hypertension
- cardiac failure
- AML
What is the therapy for PMF?
Symptom based
hematopoietic stem cell transplant
What is essential thrombocytothemia?
- ET is neoplastic stem cell disorder with proliferation of megakaryocytes
- platelet count> 450,000/uL atypical platelet morphology
- normocellular to occasionally hypercellularity BM with abnormal megakaryocytes

What are these pictures showing?

large/giant hypogranular platelets
What are 2 clinical findings for ET? What mutation is seen? What is the typical life expectancy ?
- Clinical findings:
- Bleeding or thrombosis, splenomegaly
- JAK2 mutation in 50-60% of pts. or CALR, MPL
- Most patients: normal life expectancy
- treatment: aspirin or cytoreductive therapy (hydroxyurea)
What are Myelodysplastic syndromes (MDS)
- clonal hematopoietic stem cell disorders
- cytopenias, dysplasia (one or more myeloid cell lineages) ineffective hematopoiesis, and increased risk of development of AML
- ENhanced apoptosis contributes to cytopenias
- myeloblasts < 20% (PB and BM)
What are the laboratory findings for MDS?
- cytopenias (uni-, bi- or pancytopenia)
- leukoerythroblastic reaction
- dysplastic features
- hypercellular BM
- ring sideroblasts
- increased myeloblasts
What is dysplasia? What is the difference between normal neutrophils and dysplastic neutrophils?
abnormal morphology!
- Normal neutrophils
- segmented nucleus (~3)
- granular cytoplasm
- Dysplastic neutrophils
- hyposegmented nucleus
- hypogranular cytoplasm

What is the difference between normal platelets and dysplastic platelets?
- Normal
- small size
- normal granulation
- Dysplastic platelets
- large size
- hypogranular

What is this?

Dysplastic erythroid precursor
What is this?

dysplastic megakaryocyte
*simplified nuclei with a single lobe
What is shown in these images?

ringed sideroblasts!
they have chunky granules that surround the nucleus
associated with MDS
What is a monosomy 7 mutation associated with?

MDS!
What is the clinical course of MDS?
- who gets it
- what are symptoms
- medan survivial
- what does it progress to and what causes mortality
- elderly individuals (50-80)
- weakness, infections, hemorrhage or asymptomatic
- median survivial: 9-29 months, t-MDS: 4-8 months
- progression to AML in ~30% of cases
- Mortality: infection, bleeding (due to cytopenias)
What is the therapy for MDS?
- supportive: blood products, antibiotics, growth factors
- hypomethylating agents: not curative (decitabine, azacitidine)
- allogenic stem cell transplant
What is acute leukemia and what are the 2 categories?
Neoplastic proliferation of imature cells (blasts) recapitulating progenitor cells of the hematopoietic system
two main categories: Myelobastic (myeloid)-AML and Lymphoblastic (lymphoid)-ALL
What is the difference between acute and chronic leukemia? How long do people typical live without treatment?
Acute Leukemias:
- Immature cells (blasts)
- untreated natural history of weeks to months
Chronic leukemias ( and MPNs)
- Mature cells
- untreated natural history of months to years
Who typically gets AML
3 cases/100,000 per year
primarily adults, median age is 60 yrs. It is 80-90% of acute leukemias in adults
equal male to female ratio!
Who typically gets ALL?
- most common in children, but may be seen in adults (75% under 18yo)
- 1.4 cases/100,000 ppl
- M:F= 1.4:1
What are therapeutics for ALL and AML based on?
morphology, cytochemistry, immunophenotyping, genetics
AML
- blast size
- chromatin
- nucleoli
- cytoplasm
- auer rods
- myelodysplasia
AML
- blast size: large and uniform
- chromatin: finely dispersed
- nucleoli: 1 to 4 often prominent
- cytoplasm:moderately abundant granules often present
- auer rods: 60-70% of cases
- myelodysplasia: often present

ALL
- blast size
- chromatin
- nucleoli
- cytoplasm
- auer rods
- myelodysplasia
ALL
- blast size: small to medium; variable
- chromatin: coarse
- nucleoli: absent or 1 or 2; indistinct
- cytoplasm: scant to moderate granules lacking
- auer rods: absent
- myelodysplasia: absent

What is a cytochemical stain? Which 2 are the most useful?
they exploit the presence of intracellular enzymes that produce a colored product

Myeloperoxidase (MPO): myeloblasts
Non-specific esterase (NSE): monocytic blasts in AMLs with monocyte differentiation
What are the 4 roles of immunophenotyping?
- differentiates ALL from AML
- distinguishes B-ALL from T-ALL
- Identifies subtypes of AML (megakaryocytic, monocytic etc)
- treatment and prognostic groups determined partly by immunophenotype
*using antibodies specific to antigens on the cells. they are used in situ (in tissue sections) or to bind to single cells in flow cytometry

What are the lymphoid antigens? which are associated with T lineage vs B lineage?
- T lineage: CD1a, CD2, CD3, CD4, CD5, CD7, CD8
- B lineage: CD19, CD20, CD22
What are the generic myeloid anitgens?
CD13, CD15, CD33, CD117, MPO
What are the immunophenotypic markers of immaturity? What diseases are they seen in?
CD34 (AML and ALL)
TdT (mostly seen in ALL)
CD117 (AML)
CD1a (restricted to immature T cells)
What do we use cytogenetics for?
classification: AML vs ALL associcated abnormalities
subgroups of both AML and ALL are defined by cytogenetic abnormalities (biologic and prognostic)
What is AML?
Heterogenous set of disorders with a generally poor outcome? (25% long-term survival)
Systemic neoplasms of myeloid progenitor cells that primarily involve blood and bone marrow but may involve extramedullary sites
What are the clinical features associated with AML?
- Generally related to cytopenias (weakness, fatigue, petechiae, infections)
- less common findings ( organomegaly, lymphadenopathy, infiltration of other extramedullary tissues)
- coagulopathy in specific variants
What are the general pathologic features that are required to call something Acute leukemia?
- >20% myeloid blasts in blood or marrow
- BM usually hypercellular
- variable blast morphology, depending on sub-type
- maturation may be towards any of the myeloid lineages (sometimes more than one)
- Granulocytes, Monocytes, Megakaryocytes, Erythroid precurosors
What is this red line pointing to?

The Auer rods found in AML!
What are the 2 hematologic features of AML?
Severe leukopenia to marked leukocytosis
anemia and thrombocytopenia
What are the 4 WHO classification of AML?
AML with recurrent cytogenetic abnormalities
AML with myelodysplasia-associated changes
AML, therapy related (and t-MDS, t-MDS/MPN)
AML not otherwise specified
What happens in AML with recurrent cytogenetic abnormalities (“de novo” AML)?
generally reciprocal translocations
generally flat incidence rate over differenet age groups
distinctive morphologic features
no antecedent myelodysplastic syndrome
generally favorable prognosis
What is another name for AML with 1(15;17) PML-RARA? hat is the morphology?
- “Acute promyelocytic leukemia” and is 5-8% of AMLs,
- Morphology:
- generally hypergranular (minority of cases “microgranular”)
- reniform/bilobed nuclei
- singel cells with mulitple Auer rods
- usually Leukopenic (low WBC count) but microgranular variant often manifests leukocytosis
*
What condition is associalted with APL? What therapy does it respond to?
Frequent DIC at diagnosis causes early morbidity and mortality
responds to all-trans retinoic acid (ATRA)

What does a t(15;17) produce? How do we fix it?
produces PML-RRARa fusion gene
retinoic acid is important for myeloid maturation (disruption of receptor produces maturation arrest at the promyelocyte stage)
all-trans retinoic acid (ATRA) administration overcomes blcok and essentially matures the cells and quickly corrects the coagulopathy
APL is only AML that is responsive to ATRA
What is AML with Myelodysplasia related changes likely related biologically to? What is the prognosis?
- Likely related biologically to MDS, and may be antecedent MDS
- MDS-type cytogenic abnormalities (complex karyotypes, losses of chromosomes or parts of chormosomes)
- increasing incidence with age
- prominent multilineage dysplasia
- poor prognosis

What is Therapy-related AML (t-AML) (and t-MDS, t-MDS/MPN) ?
AML (or MDS, MDS/MPN) occuring in patients previously treated with chemotherpay and/or radiotherapy
Alkylating agent related
Topoisomerase
ionizing radiation (involving bone marrow)
other agents
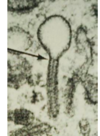
What is langerhans histiocytosis?
a histiocytic condition that stains for CD1a and langerin
distinct clinicopathologic entities
Birbeck granules (“tennis racket” appearance on EM)
BRAF mutations

What are the 3 kinds of langerhans cell histiocytosis?
- Multifocal multisystem
- often >2yrs old
- aggressive
- Unifocal/unisystem
- often involve bones
- treatable; sometimes spontaneous regression
- Pulmonary
- associated with smoking, may regress after quitting
WHat is Hemophagocytic lymphohistiocytosis/ hemophagocytic syndrome (hyperinflammatory condition)?
- non-neoplastic but aggressive (often fatal) clinical pathologic syndrome
- uncontrolled systemic activation of macrophages, cytotoxc T cells, systemic inflammation
- Primary (genetic) HLH: defects in genes involve in T/NK cell cytotoxic granule formation/relase (ie perforin)
- Secondary HLH: Infection (EBV), Neoplasms (lymphomas)
What are the 8 diagnostic criteria for hemophagocytic lymphohistiocytosis/ hemophagocytic syndrome (hyperinflammatory condition)?
1 genetic link or 5 out of the 8 below criteria
- Fever (>38.5 for 7 days)
- Splenomegaly (palpable spleen >3cm below costal margin)
- Cytopenias involving> 2 cell lines (Hb<9, ANC <100, platelets<100,000)
- Hypertriglyceridemia or hypofinbrinogenemia (fasting TG>2 mmol/L, fibrinogen<1.5g/L) *or 3SD away from typical for age
- Low or absent Natural Killer Cell activity
- Serum ferritin >500uG/L (usually 1000s)
- Elevated soluble interleukin-2 (CD25) levels (>2400U/mL or high for age)
