10b- Intro to Hemostasis- Bleeding Disorders Flashcards
(42 cards)
What are 5 defects in primary hemostasis?
- skin and mucosal membrane hemorrhages are common (eg petechiae, epistaxis, menorrhagia)
- Vascular abnormalities (eg Ehlers-Danlos)
- Platelet function disorders
- von Willebrand disease
What are 3 defects in secondary hemostasis?
- bleeds into soft tissues or joint
- clotting factor deficiencies
- hemophilia
- liver disease
- exogenous
- anticoagulants
What is thrombocytopenia? What 4 things are also on the differential diagnosis?
- usually defined as <100,000/uL
- Differential
- decreased bone marrow: aplastic anemia
- increased destruction (immune mediated, non-immune mediated)
- dilutional: decreased platelets bc increased fluid
- sequestration: big spleen harbors platelets
Thrombocytopenia is decreased platelets due to decreased _______
What 7 things cause thrombocytopenia?
Thrombocytopenia is decreased platelet production due to decreased megakaryocytes
- aplastic anemia
- vitamin B12/folate deficiency
- leukemia, lymphoma, MDS
- metastatic carcinoma
- alcohol, toxins
- drugs
- infections

Thrombocytopenia caused by increased destruction can be immune mediated or non-immune mediated? What are 4 examples of immune mediated thrombocytopenia?
immune thrombocytopenia purpura (ITP)
Heparin-induced thrombocytopenia (HIT)
Transfusion/preganncy-associated allo-immune thrombocytopenia
drug (quinine, vancomycin)
What are 4 non-immune mediated causes of thrombocytopenia?
Disseminated intravascular coagulation (DIC)
Thrombotic thrombocytopenic purpura (TTP)
Hemolytic uremic syndrome (HUS)
Drug
WHat is Immunt Thrombocytopenia Purpura (ITP)
Caused by autoantibodies made against PLT antigens (GpIIb/IIIa, GpIb)
destruction of PLTs occurs by phagocytosis, Fc receptor mediated, loction is the spleen
What are the 2 categories of immune thrombocytopenia purpura (ITP)
- Primary: unknown etiologies
- Secondary: diseases we can identify
- lupus
- leukemia/lymphoma (eg CLL/SLL)
- Drugs
- Viruses (eg HIV, Hepatitis)
What are the clinical features of Immune Thrombocytopenia Purpura (ITP)? WHat are the 2 variant/the difference between them?
- skin, mucosal bleeds
- normal size spleen
- variants:
- Acute: self limited (< 6 months, post-viral, children)
- Chronic: adults (females), persistent>6 months
What are the pathologic findings of ITP?
Thrombocytopenia (often quite low), large platelets (circled)
normal to increased megakaryocytes in marrow
white pulp expansion

How do we diagnose ITP?
presumptive
iagnosis of exclusion
no diagnostic test
normal clotting tests
bone marrow not required
How do we treat ITP?
- May not be needed in children with self-limited disease
- immunosuppresant therapy
- steroids, intravenous immunoglobin (IVIG)
- Anti-CD20 antibody
- Thrombopoietin agonist
- Splenectomy
What happens in drug-induced thrombocytopenia?
drug binds to IIa/IIIb to elicit antibody reaction.Phagocytic cells then localize to the antibody and destroy the platelet

What is dilutional thrombocytopenia? Sequestration thrombocytopenia?
Dilution: Massive transfusion of trauma patients
Sequestration: Seen with hypersplenism, increased storage fo Platelelets
What are platelet function disorders? List 5
heterogenous group of inherites and acquired, qulitative disorders with variable clinical presentation
Bernard Soullier
Glanzmann’s thrombasthenia
Storage pool disease
Aspirin
Uremia
What is the pathophysiology of Bernard Soullier?
Inherited vs acquired
Clinically severe or variable?

Gp1b deficiency on platelet memebrane
inherited
severe
What s the pathophysiology of Glanzmann’s thrombasthenia?
Inherited or acquired
severe or variable
GpIIb/IIIa deficiency on platelet memebrane
inherited
severe

What is the pathophysiology of storage pool disease?
inherited or acquired
clinically severe or variably
granule, transit defect
inherited
variable

What is the pahtology of aspirin platelet dysfunction? inherited or acquired? clinically sever or variable?
Cox inhibition
acquired
variable

What is the most common inherited bleeding disorder? How is it inherited and how does that relate to severity of disease?
vonWillebrand disease
- usually autosomal dominant, incidence may be underestimated given mild presentation. rarely acquired
- clinically heterozygous
- mild, revealed with surgery, dental procedure, trauma
- moderate to severe, menorrhagia, spontaneous bleeding
- dependent on clinical type
Where is vW factor produced? What is the function? What is the structure of vWF
- produced by endothelial cells, megakaryocytes/PLTs
- Function:
- When circulating: stabilize F8 (T1/2=2.4 hrs free to 12 hours bound)
- Fixed: adhere to collagen and platelets through Gp1b
- Structure: multimers-metalloproteases cleave freshly cut vWF into multimers. there should be equal amount of all of the multimers

What are the 3 types of vWD? What are the characteristics of each of them?
- Type 1: Quantitative: most common A/D
- clinically heterogenous usually mild (how much factor they have determines how severe the disease is)
- Type 2: Qualitative
- 2A, 2B, 2M, 2N is A/R; others are usually A/D
- moderate to severe bleeding (They make vWF but it is dysfunctional)
- Complete absence (Quantitative)
- Rare, A/R
- severe bleeding disorder, mimichking heterophilia
How do we treat vWD?
desmopressin stimulates vWF release
factor concentrates
anti-fibrinolytics
oral concentration (to stop menorrhagia)
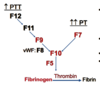
What are the laboratory tests used to screen for vWD and what would we expect the results to be? What are the confirmatory studies? (sorry this is a long card)
- Screen
- CBC (normal PLT count)
- PT (normal), aPTT (normal or prolonged)
- vWF antigen levels
- vWF activity (ristocetin)
- Factor 8 activity
- Confirmatory studies
- multimer electrophoresis
- specific biinding assays
- platetlet aggregation