11/19- Disorders of Connective Tissue- Skeletal Flashcards
Describe collagen biochemically
- How many types
- Mutations cause what
- __ bound molecules
- Where are precursor chains made
- Other parts of production
- Collagen bio-synthesis is complex
- More than 16 different types, very abundant in connective tissue
- Mutations in different collagen chains will lead to different diseases where those chains are mostly abundant
- Triple bound molecules, individual precursor chains are synthesized in membrane bound polyribosomes
- Hydroxylated and glycosylated in the rough endoplasmic reticulum (RER)
- Transport, extrusion and proteolysis to remove carboxy (start) and amino (end) terminal propeptide extensions
What are the different types of collagen and where are they found?
- Components
- Diseases associated
Collagen type I is the one we’ll focus on
- a1, a2, and a3
- Found in skin, tendon, bone, and arteries
- Associated with Osteogenesis Imperfecta
Other fun facts:
- Collagen type 2: cartilage and vitreous humor
- Collagen type 3: EDS type IV
- Collagen type 4: found in basal lamina
- Collagen type 5: EDS type I

Type I procollagen is made of what?
- What chromosomes are involved
- 2 pro-alpha 1 chains (chr 17)
- 1 pro-alpha 2 chain (chr 7)
Describe the structure of type I procollagen
- Triple helical structure arranged of tandem Gly-X-Y repeats
- X = proline
- Y = hydroxyproline or hydroxylysine
What is Osteogenesis Imperfecta
- Inheritance pattern
- Genes involved
- Phenotypes
Disorders of collagen and collagen chaperon molecules (post-translation: hydroxylation).
- Most common forms are autosomal dominant.
- Rare recessive: CRTAP, FKBP10, LEPRE-1,PPIB, SERPINF1, SERPINH1, SP7.
- Quantitative defects are associated with milder phenotypes while qualitative defects are more severe.
Describe the basics of OI type I
- Incidence
- Protein involved
- Phenotype
Type I = mild form
- 1/15-20,000
- Type I collagen
- Phenotype
- Multiple recurrent fractures (common an ambulation; steady rate of fractures through childhood and then decrease after puberty; start up again after menopause in women and 60-80 yo in men)
- Normal stature
- Little or no bone deformity
- Blue sclera
- Hearing loss in 50%
- Quantitative defect
What are the type of mutations contributing to OI type I?
Pro-a1 null mutations
- Haploinsufficiency of type I collagen (quantitative defect)
- 1/2 the normal amount of normal type I collagen
What is seen here? What causes it?

Blue/grey sclera in OI type I
- Due to thinning of sclera with color of vessels showing through
What is seen here?

Compression fractures
- Bioconcave appearance of vertebrae (fish-shaped vertebrae)
Describe the severities of type II, III, and IV OI?
- Type II (fatal): lethal in the neonatal period
- Type III (deforming): severe and progressive deformity at birth
- Type IV: mild to moderate bone deformity and variable short stature, common dentinogenesis imperfecta (DI), variable sclerae, and hearing loss
As opposed to type I OI, types II-IV are ______ defects
As opposed to type I OI, types II-IV are qualtitative defects
Describe type II OI
- Incidence
- Gene mutation/consequences
- Prognosis
- Phenotype
- Perinatal, lethal
- Affects 1/20-60,000 (much rarer than type I)
- Mutations in COL1A1
- Glycine substitutions and mutations in the C-terminal pro-peptide (where the protein starts)
- Phenotype: Minimal calvarial mineralization, beaded ribs, compressed femurs, long bones bowing, platyspondyly, small thoracic cage (pulmonary hypoplasia)
- Ultimate cause of death is pulmonary hypoplasia
What is seen here?

OI type II (lethal)
Describe type III OI
- Phenotype
- Qualitative collagen defect.
- Very short stature and bone deformities at birth due to in utero fractures.
- Variable sclerae, dentinogenesis imperfecta (DI), hearing loss.
- Recurrent fractures with minimal trauma + pain.
- Severe deformities with ambulation restriction.
- Adult height 3 ft - 4 ft.
- Pulmonary insufficiency due to severe kyphoscoliosis.
What is seen here?

OI Type III (deforming type)
What is seen here?
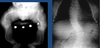
OI type III (deforming type)
Describe the stochiometric effect going on with Pro-a1 in OI types II, III, and IV
- Decreased rate of triple helix formation
- Increased post-translational modification of NH2 terminal to mutation
- Decreased secretion and increased degradation
- Defective collagen fibrils
- Poor mineralization (in bone)
Describe the stochiometric effect going on with Pro-a2 in OI types II, III, and IV
- Biochemical abnormalities similar to the above (a1) but may be less severe
- Phenotype depends on substitution
Why are qualitative defects worse than quantitative?
- Up to 75% of collagen molecules are abnormal
- Abnormal trimers (greater severity than quantitative defects)
- Exporting abnormalities = protein suicide (dominant negative)
Which types of OI are AD?
All of them (I-IV)
What types of mutations are more severe in OI?
“Subtle” point mutations (glycine substitutions) are more severe than large rearrangements which results in null alleles
- Phenotypic gradient from carboxy-terminal to amino-terminal mutations
____ have a ___ effect on normal collagen chains
Mutant collagen chains have a dominant negative effect on normal collagen chains
Mutations in ___affect __% of collagen trimers while _____ mutations affect __% of the trimers
Mutations in COL1A1 affect 75% of collagen trimers while COL1A2 mutations affect 50% of the trimers
What are treatments in OI?
- Supportive
- Interventional
Supportive
- Orthopedic management: intramedullary rods, external braces, splints, castings
- Hearing aids
Interventional
- Infusion of biphosphonates: pamidronate (Aredia®), zoledronic acid (Zometa®)
Describe the process of bone renewal and remodeling
- Regulation
- Effect of homones
- Process that continues throughout life
- Regulated by hormones and cytokines
- Resorption of bone and deposition work in parallel (coupling)
- Lower estrogen levels cause an increment in exchange with coupling dissociation => bone loss

What is the basic mechanism behind bisphosphanate function?
Inhibit osteoclastic activity and induce apoptosis
What are the 2 types of bisphosphanates?
- Pyrophosphate analogs: Clodronate, etidronate
- Nitrogen Analogs: Pamidronate, alendronate, risendronate, ibandronate, zoledronate
Describe Pamidronate and Zoledronic acid
- What type of bisphosphonate
- Benefits
- Side effects
- Nitrogen analogs
Benefits:
- Reduces pain caused by osteoporosis
- Increases the bone density
- Restores the shape integrity and height of the vertebral bodies
Side effects: osteonecrosis of the jaw (due to interference in angiogenesis)
Describe the formation of skeletal elements (the developmental process and types of bone)
- Intramembranous ossification gives rise to the flat bones that comprise the cranium and medial clavicles
- Ossification is accomplished directly.
- Endochondral ossification gives rise to long bones (appendicular skeleton, facial bones, vertebrae, and the lateral medial clavicles).
What are the subtypes of micromelic (short) limbs?
- Rhizomelic: short proximal limb
- Mesomelic: short distal limb
- Acromelic: short appendage (hand, foot)

What are the most common skeletal dysplasias?
- Prevalence
- Thanatophoric dysplasia (1/10K)
- Achondroplasia (1/20K)
- Achondrogenesis (1/40K)
- OI type II (1/60K)
Many more…
Describe the genetics of Achondroplasia
- Inheritance pattern
- How many de novo
- Associated with what
- Gene/chromosome involved
- Incidence
- Autosomal dominant
- 80% are new mutations
- Advanced paternal age
- Incidence 1/20,000
- Gene mutated is FGFR3 located in 4p16.3
- This is the most commonly mutate gene in humans (?)
- 95% of cases change Glycine to Arginine in AA 380
What are FGFRs?
- Which is affected in achondroplasia
4 tyrosine kinase molecules that bind and are activated by most of the FGFs molecules (FGFR 1, 2, 3, and 4)
- FGFR 3 is the one involved in Achondroplasia (ligands unknown)
What happens when FGF ligands bind FGFR3?
- Binding of FGF ligands to FGFR3 causes monomers to dimerize
- Activates TK
- Phosphorylation of tyrosine residues serve as docking sites for signaling molecules recruited for the receptor
- MAP and STAT kinase signaling pathways relevant to inhibition of chondrocyte proliferation
What happens to FGFR3 in Achondroplasia?
- Consequences?
- This mutation activates FGFR3 in the absence of ligand
- Function: normal effect of signaling FGFR3 limits chondrocyte proliferation and differentiation within the epiphyseal growth plates and cause achondroplasia - Knockout FGFR3 mice have “longer” bones
- FGFR3 appears to restrain and inhibit bone growth.
Achondroplasia appears to be the result of “a gain of function mutation” suggesting that receptor downregulates FGF signal transduction
What are clinical features of Achondroplasia?
- Macrocephaly, frontal bossing, midface hypoplasia
- Short stature, rhizomelic shortening of the extremities
- Trident hands
- Short stature
- Lumbar lordosis
- Brachydactyly on exam and X-ray

What is Thanatophoric Dysplasia?
- Incidence
- Genetics
- Phenotype
- “Thanatos”: death-seeking
- Incidence: 1/10,000
- Similar to homozgyous achondroplasia
- Mutations in FGFR3:
- IgII/III like domains for TD type I
- TK domain in TD type II
- Short femurs, flat vertebral bodies, short ribs

What is the treatment for Thanatophoric Dysplasia?
- Conservative symptomatic management: suboccipital craniectomy for foramen magnum compression, spinal release, lower extremities osteotomies in case of bowing.
- Experimental: Modified C-Natriuretic peptide (CNP)
What is CNP? How does it work?
Modified C-Natriuretic peptide (CNP) used to treat Thanatophoric Dysplasia
- CNP influences body fluid homeostasis and blood pressure control
- CNP added to organ cultures of mouse tibias increases their length
- Histological exams reveal the CNP increases the height and hypertrophic condrocyte zones in tibial epiphyses
- CNP rescues achondroplasia phenotype in mice
- CNP can rescue achondroplasia in achondroplasia mouse models and promotes long bone growth in normal monkeys
- Current clinical trials (Phase II) are underway to assess modified CNP in a group of young children with achondroplasia
