11/10- Cancer Genetics Flashcards
Brief history of oncogenes
- First cloned using viruses that conveyed cancer to recipient animals by infection of viral DNA or RNA.
- Then cloned by transformation/transfection assays of genomic DNA from tumor cells places into mouse fibroblasts which caused the recipients cells to grow abnormally – called “transformation”.
- Sequencing of transformed cells revealed a human gene from the tumor with a single point mutation compared with normal human DNA.
What is a “proto-oncogene”? “Oncogene”?
- Proto-oncogene: normal cellular counterpart
- Oncogene: mutated form found in the tumor
(However, some papers refer to activated oncogenes as the mutated form)
Oncogenes have diverse functions. What are some of them?
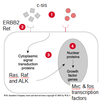
Functions (start outside the nucleus)
1. Growth factors/ligands: c-Sis.
2. Cell Surface Receptors: large class of proto-oncogenes which have been important as targets of new cancer drugs:
- Tyrosine kinase receptors, e.g., c-kit, ERBB2, RET.
3. Signal Transduction Molecules: another large class that are very frequently mutated and are now drug targets.
- ALK, RAF, H-. K- and N-RAS commonly mutated.
4a. Cell cycle regulatory molecules
4b. Transcription factors
5. Regulators of apoptosis (programmed cell death)
Reflects that many different ways to turn a normal cell into a tumor cell
What are cell cycle regulator molecules with relation to oncogene function?
- Describe activation
The most important regulatory molecules in the eukaryotic cell cycle are the Cyclin Dependent Kinases, CDC2, CDK2, CDK4
- To be active the Cdk must be bound to another protein called a cyclin.
- Activation of cyclin D1 or CKD4 – occurs in many tumors and drives the cells through the cycle to aid in proliferation.
What are transcription factors with relation to oncogene function?
c-myc - overexpression expedites the G1 - S transition, makes cells resistant to differentiation factors.
- Activation of transcription factors is a major feature of leukemia and pediatric solid tumors.
- Alterations in miRNAs in tumors can alter the expression of large gene sets at the same time.
What is an example of regulators of programmed cell death (apoptosis) being dysfunctional in cancer?
BCL-2 is a molecule which prevents normal programmed cell death from occurring and is upregulated in follicular lymphoma.
- BCl-2 inhibits ICE protease that cause DNA fragmentation and apoptosis
What is seen here?
- Mechanism/oncogene?

Follicular Lymphoma: contains too many well differentiated, normal looking lymphocytes.
- Results from anti-apoptotic effects of BCL-2 overexpression, which itself results from t(14;18)(q32;q21) translocation
What are the origins of mutations in oncogenes?
- The types of mutations differ between those found in oncogenes and tumor suppressor genes.
- Mutations in oncogenes activate the gene product (missense) or cause the gene to be misexpressed (translocations) or overexpressed (amplification)
- Mutations in oncogenes are frequently somatic, e.g., found in the tumor but not in matched normal DNA from the patient.
- You see one activated oncogene and one normal proto-oncogene allele in the tumor.
- Rare syndromes that result from inheriting an activated oncogene.
What are examples of inherited oncogene mutations?
- RET
- MET
- HRAS
- KRAS
- ALK
Describe the cancers/syndromes behind the inherited oncogene mutations
- RET
- MET
- HRAS
- KRAS
- ALK
- RET: mutations cause multiple endocrine neoplasia type 2 (esp medullary thyroid cancer)
- MET: mutations result in hereditary papillary renal cell cancer
- HRAS: mutations result in Costello syndrome with skeletal abnormalities, developmental delay, bladder cancer, neuroblastoma, and rhabdomyosarcoma
- KRAS: mutations result in Cardi-Facio-Cutaneous syndrome; no known cancer phenotype yet
- ALK: mutations responsible for hereditary neuroblastoma
What are the most common types of mutations seen in proto-oncogenes that activate them?
- Point mutations
Describe how point mutations contribute to development of oncogenes. Examples
SPECIFIC POINT MUTATIONS which alter the normal activity of a protein to make it more tumorigenic.
- Normal RAS protein has an inactive state when bound to GDP.
- RAS missense mutation found in tumors results blocks GTPase yielding constitutively active RAS protein.
- RET can have single missense mutations in cysteine residues which activate the receptor in the absence of ligand.
Somatic RAS mutations are very common base substitutions in human tumors.
What are the different RAS mutations? Cancers that prefer each type?
- HRAS
- Bladder cancer
- KRAS
- Pancreas cancer
- Colon cancer
- Lung cancer
- Uterus cancer
- NRAS
- Leukemia
- ?RAS
Identification of somatic mutations in tumors is becoming increasingly important in making treatment decisions for patients.
- Missense mutations in the ___ oncogene in tumor samples predict the response of ____ cancer patients to _____
Missense mutations in the EGFR oncogene in tumor samples predict the response of lung cancer patients to Gefitinib
Missense mutations in ____ convey sensitivity to _______ in ______ cancer
Missense mutations in RET convey sensitivity to Vandetanib in medullary thyroid cancer
- The availability of biopsy tissue for molecular analyses is increasing important for treatment decisions
How does amplification play a role in cancer development? Examples?
Amplification results in increased copy number of an oncogene and presumed increased expression of the gene
What are examples of cancers that make use of amplification as a mechanism for oncogenic potential/
- MYCN (N-myc) amplification is frequently found in neuroblastoma cells and correlates with prognosis.
- Treatment decision at diagnosis requires assessing the MYCN status of neuroblastoma.
- MYCC (c-myc) amplification occurs in about 40% of human breast cancers but is not currently used for treatment decisions.
MYCN amplification in neuroblastoma is associated with what?
Advanced stage and poor prognosis
Describe how translocations/rearrangements contribute to oncogenic potential
Chromosomal rearrangements, in particular, translocations- cause the gene to be abnormally expressed by bringing together two different chromosome fragments.
- A hallmark of hematopoietic malignancies.
- Specific translocations now also be found in many solid tumors.
- Because the proteins created by translocations are unique found in the tumor cells they are a major target of new drug development.
Imprecise translocations result in what?
an oncogene being moved to the proximity of a transcriptionally active gene.
Precise translocations result in what?
The precise joining of two genes to make a novel fusion gene:
- The 5’ end of the gene controls the expression pattern +/- functional domains and the 3’ end of the gene often controls function.
- The BCR and ABL genes form a fusion gene encoded by the Philadelphia chromosome of CML.
- Produces a novel kinase which is the target of a specific antitumor agent, Imitamib/Gleevac
Describe the _____ (precise/imprecise) translocation in Burkitt’s lymphoma
Imprecise!
- Activation of c-Myc oncogene by juxtaposition of c-Myc with the Immunoglobulin locus in lymphoid cells in Burkitt’s Lymphoma – no unique fusion protein is made
Describe the __ (precise/imprecise) translocation resulting BCR-ABL kinase
Precise!
- Derivative of chromosome 22 with a bit of 9 stuck on the end (der (22) Ph)
What is the Philadelphia chromosome?
22: 9 chromosome translocation
- Results in BCR-ABL kinase
What cancer is Philadelphia seen in?
CML- chronic myelogenous leukemia
The presents of BCR-ABL gene means what for the treatment of that cancer?
- Could develop of a specific inhibitor against the BCR-Abl protein
- Resulted in improved treatment of Ph+ leukemia (CML and ALL) with improved survival.
- Now also used for tumors which have activation of other related kinases, like gastrointestinal stromal tumors (GIST) with activation of c-Kit kinase
Translocations/fusions have since been found in many diverse cancer types. What other cancers commonly involve them?
- Translocations: ~80% of prostate cancer; most common is androgen responsive TMPRSS2 gene and the ERG oncogene.
- Translocation: activation of ALK kinase in anaplastic lymphoma.
- Deletions between gene: P2RY8 promoter and exon 1 are fused to CRLF2 coding region (growth factor) in high risk ALL and Down Syndrome-ALL.
What is the Chr2 inversion? what does it result in?
Chr 2 inversion -> EML4-ALK fusion
- Inversion event is seen in ~3% of non-small cell lung cancer.
- Results in activation of ALK because the EML4 domain aids in heterodimerization
What are the therapeutic implications of Chr 2 inversion -> EML4-ALK fusion
- Early trials supported the use of ALK inhibitors for the treatment of EML4-ALK–positive NSCLC
- Crizotinib approved for EML4-ALK rearranged NSCLCs
- Targeting of activated ALK may be successful therapeutic approach to diverse tumors types.
- Pediatric Phase 1 trial of crizotinib demonstrated activity against tumors with rearranged ALK.
- Including ALK translocations, inversions
- However, less clear efficacy for tumors with missense mutations
What are the specific kinase alterations in cancers:
- JAK2
- ALK
- FLT3
- EGFR
- JAK2 mutations in high-risk childhood ALL and ALL associated with Down syndrome
- ALK mutations (and amplification) in familial and sporadic neuroblastoma
- FLT3 internal tandem duplication in AML.
- EGFR missense mutations in lung cancer associated with sensitivity to tyrosine kinase inhibitors (gefitinib)
- Secondary somatic mutations V843I or T790M then associated with resistance to same class of drugs
What are the specific kinase alterations in cancers:
- Childhood ALL and Downs-associated
- Neuroblastomas
- AML
- Lung cancer sensitive to Gefitinib
- JAK2 mutations in high-risk childhood ALL and ALL associated with Down syndrome
- ALK mutations (and amplification) in familial and sporadic neuroblastoma
- FLT3 internal tandem duplication in AML.
- EGFR missense mutations in lung cancer associated with sensitivity to tyrosine kinase inhibitors (gefitinib)
- Secondary somatic mutations V843I or T790M then associated with resistance to same class of drugs
What is the BRAF gene? Its importance?
- BRAF (B-raf) gene was found to carry a specific mutation V600E in a substantial percentage of malignant melanoma:
- Subsequently identified in many other advanced cancers and pediatric brain tumors.
- Targeted trials of BRAF inhibitors led to rapid FDA approval of several drugs for V600E mutated melanoma
- Similar mutations at orthologous position in KIT (D>V) as seen in BRAF in other tumor types.
Whole exome sequencing of high grade gliomas by Hopkins group (Will Parsons) led to discovery of gain of what?
Function mutations in IDH1/2 in astrocytomas and secondary glioblastoma multiforme.
- IDH1/2 also frequently mutated in AML
- Functional assays reveals that these are gain of function mutations which alter substrates for chromatin remodeling
What are FDA approved targeted cancer drugs (off label use in italics)
- Imatinib
- Trastuzumab
- Crizotinib
- Vermurafinib
- Erlotinib
- Vandetanib
Germline status:
- Everolimus
- Olaparib

Oncogene summary
- Activation of proto-oncogenes by mutation, amplification and translocation result in increased or altered regulation that fosters tumor cell growth and survival.
- Mutations in oncogenes are occasionally inherited
- Alterations maybe specific to the type of cancer but same gene often mutated in several different tumors
- Resulted in development of many new targeted therapies.
- Cancer genome projects seek to identify all such changes to allow for specific and less toxic treatment for a wide variety of cancers
T/F: Most cancers result from inherited predisposition to cancer
False
- For most tumor types, e.g. breast, the inherited fraction fall in the range of 5-10%.
- Several rare tumors, adrenocortical carcinoma, retinoblastoma and medullary thyroid cancer have very high inherited fraction (40-60%).
The majority of inherited cancer result from ____ mutations in _______
The majority of inherited cancer result from loss of function (LOF) mutations in tumor suppressor genes
- Rare examples of inheriting oncogenic driver mutations The risk of cancer in mutation carriers is high enough to result in specific management guidelines.
Patients with inherited chromosomal abnormalities may have an increased risk of ______. Example?
Patients with inherited chromosomal abnormalities may have an increased risk of developing cancer
- Trisomy 21 results in ~20 fold increase in leukemia and also shifts the myeloid: lymphoid ratio to 40:60.
- Not an increased risk of solid tumors in adults with DS
- Girls with mosaic Turner syndrome or gonadal dysgenesis at increased risk for gonadoblastoma.
- Correlates with presence of Y chromosome
- Children who survive trisomy 18 have a very high rate of developing Wilms tumor.
Describe the pathogenesis of leukemia in DS
- Transient myeloid proliferation (TMP)
- May or may not evolve into leukemia is seen in infants.
- Somatic GATA1 mutations found in TMP cells
- JAK2 missense mutations and CRLF2 activation due to intrachromosomal deletion found in DS-ALL

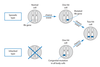
What were some early observations about Rb?
- Childhood eye cancer which can be seen in absence of family history (sporadic) or inherited in an autosomal dominant manner.
- Patients with a family history of Rb are much more likely to have bilateral tumors.
- Appeared to have earlier age of diagnosis
- Knudson reviewed 48 cases of Rb and applied statistical analysis which showed that it fit a model of only two events or “hits” being required for tumor formation

Look at this picture about the 2 hit hyopthesis
Cancer risk for BRCA1/2 mutation carriers (table)
Despite the increased risk for melanoma and pancreatic cancer.. no guidelines for screening

Summary of risk reduction strategies for mutation carriers

What is Familial Adenomatous Polyposis (FAP)?
- Autosomal dominant disorder due to mutations in APC with very high penetrance for polyps and colorectal cancer
- Adenomatous colonic polyps begin in childhood to adolescence.
- Nearly 100% lifetime colorectal cancer risk with 50% risk by age 33.
- Extracolonic features often referred to as Gardner Syndrome.
There is a rare autosomal recessive disorder (MUTYH) which gives a similar adenomatous polyposis condition

Look at this picture for the revised two-hit hypothesis!
- Your first hit coud be mutation of methylation
- From there, could have 2nd hit of LOH or methylation
- Two hits could be any combo:
- Mutation + LOH
- Mutation + methylation
- Methylation + LOH
- Biallelic methylation
- If Lynch syndrome, this whole thing would start already with 1 mutated gene

What is recommended evaluation of colon cancer patients?

Another picture summary of molecular genetic evenst in the evolution of colon cancer

What is Xeroderma Pigmentosum?
- Pattern of inheritance
- Symptoms
- Cancer risk
Autosomal recessive disorder
- Starting at ~ age 1 patients demonstrate: easy sunburn, photophobia, photosensitivity and freckling.
- Premature skin changes with wrinkling, solar keratoses, telangiectasias.
- 50 year earlier onset and 1000x relative risk of benign and malignant skin cancers.
- 5% develop melanoma

What are overall approaches to hereditary cancer testing?

