Renal Pathophysiology Flashcards
How is GFR measured?
clearance of inulin or creatinine
estimates based on serum creatinine
azotemia
accumulation of nitrogenous waste products in the blood
i.e. urea
any rise in serum BUN or creatinine above normal
uremia
clinical syndrome or symptom compelx associated with severe impariment of renal function
specific gravity of urine
lower specific gravity correlated with low osmolarity (more dilute urine)


This is an example of a urine
1: Here a white cell with red blood cells around it
When you see cells in the urine you do not know if they have come from the kidney or someplace else in the urinary tract a

WBCs and bacteria in urine

tubular epithelial cell (not round like WBC)

squamous epithelial cells - from bladder ureter or urethra NOT kidney
casts
cylindrical masses of agglutinated material
formed in distal nephron, have to come from kidney
Tamm-Horsfall mucoprotein is the major protein constituent
Hyaline, granular or cellular
Where are casts formed?
distal nephron
Tamm-Horsfall mucoprotein
major protein constituent of casts
Hyaline cast
we think the hyaline cast and granular cast are degenerated cellular casts
There is a lot of other amorphous material here

tubular epithelial cell cast
you can see the shape of the cells here are not perfectly round which you would see in a white blood cell cast

broad cast
it was formed further down in the nephron, again there are red cells around this cast

coarse granular cast
notice the granules and degenerating cells

RBC cast

WBC cast

waxy cast (probably has cholesterol)

triple phosphate crystals
often in people with UTIs

calcium oxalate crystals

On the left there are stellar and amorphous Ca Phosphate crystals
On the right Ca Oxalate crystals

cysteine crystals

uric acid crystals
What does dipstick look for?
•Dipsticks (mainly picks up albumin, may miss low molecular weight and other nonalbumin proteins)
Heat and Acetic Acid for urine test?
take a specimen of urine and heat it up and if there’s protein you will see it form at the bottom of the test tube
The test tube has been heated (left) and as it cools you see the protein on the bottom
This is with heat and acetic acid but with sulfosalicylic acid it will look very similar
We don’t do this often, usually send sample off to the lab and they can measure protein or albumin
sulfosalicylic acid test for urine
detects all proteins in the urine
Microalbuminuria - how do we test?
dipsticks not positive until rel high
to find smaller amounts - use direct measrements of albumin secretion
microalbumin - to - creatinine ratio!
How do you determine the type of protein in the urine?
protein electrophoresis
glomerular proteinuria
increase in permeabilty of glomerular capillary wall leads to increased glomerular filtration of protein
tubular proteinuria
impaired reabsorption of normally filtered proteins
overflow proteinuria
increased production of smaller proteins in multiple myeloma and ther plasma cell issues
nephrotic syndrome
massive loss of normal serum proteins in the urine
- heavy proteinuria (>3.5)
- hypoalbuminemia
- edema
- hyperlipidemia
- sometimes HTN
hypoalbuminemia in nephrotic
urinary loss of protein
liver is making more but can’t keep up with loss

Why edema in nephrotic syndrome?
overfill hypothesis
glomerular disease/tubular inflammation leads to increased renal sodium retention (reabsorb mostly in collecting tubules
Why is there no hypertension in nephrotic syndrome?
Na retention USUALLY results in hypertension but nephrotic patients to not
MAY be secondary to hypoalbuminemia
Why hyperlipidemia in nephrotic syndrome?
elevated cholesterol, TG, phospholipids
low plasma albumin?
increased lipoprotein synthesis
lipiduria in nephrotic syndrome
oval fat bodies (tubular cells w fat drops)
maltese crosses (fat drops under polarized light)
on urinalysis

oval fat bodies in neprhotic synd urine
1: oval fat body (tubular cell that is filled with fat)

maltese crosses - polarized light on fat
lipid in urine in nephrotic
If you think a patient has nephrotic syndrome it is important to look at the urine for oval fat bodies and then look under polarized light
thromboembolic events
in nephrotic syndrome!
hypercoagulable state
DVT and renal vein thrombosis
Minimal Change Clinical Picture
acute onset
variable fluid retention
HTN infrequent
renal function is normal
EDEMA and protein in the urine!
urinalysis in minimal change disease
proteinuria (ALBUMIN - selective)
oval fat bodies
few cells
treatment for minimal change
high dose steroids - usually remission in 2-4 wks
membranous nephropathy presentation
insidious - asymptomatic proteinuria or microscopic hematuria
urinalysis in membranous
massive proteinuria (non selective - not just albumin)
HTN and azotemia if late
acute glomerulaonephritis presentation
follows GSA - pharyngitis or skin
gross hematuria and oligouria
edema and pulmonary congestion
flank pain
hypertension
when do you see congested circulation?
nephritic!!
renal retention of salt and water
decreased urine output
dyspnea, orthopnea, cardiomegaly, rales, gallop
urinalysis in acute glomerulonephritis
GAS rxn
hematuria (coca cola)
RBCs, RBC casts
prteinuria (low)
low urine sodium (retaining, vol overload)
very concentrated urine
treatment of acute glomerulonephritis
treat HTN
manage fluids and electrolytes
treat renal failure/dialysis
Hematuria in which syndromes/
gross - nephritic only
microscopic - sometimes nephrotic, always nephritic
hypertension in which syndromes
sometimes in nephrotic, always in nephritic
decreased GFR in which syndromes?
sometimes nephrotic
always nephritic
congestion in which syndromes
only nephritic
hypoalbuminemia in which syndromes
always nephrotic
rarely nephritic
urinalysis in UTI
pyuria and WBC casts
bacteria
urinalysis in pyelonephritis
WBCs and WBC casts
Urinalysis in acute interstitial nephritis
eosinophils
granular or WBC casts
systemic glomerulopathies
nephrotic
diabetes, amyloid
primary glomerulopathies
nephrotic
minimal change
fsgs
membranous
systemic nephritis
SLE
Endocarditis
MPGN
ANCA
kidney only nephritic
post infectious
IgA
congenital nephrotic syndrome of the newborn
finnish
severe NS at birth - all ESKD
need dialysis and transplant
because mutation in nephrin (in the podocyte slit diapragm)
how we learned about it!
secondary causes of minimal change
malignancy (Hodgkin and non-hodgkin lymphoma
drugs (NSAID, lithium, rifampin)
Infections (syphilis, malaria)
clinical presentation of minimal change
mostly children
explosive onset - edema, hypoalbuminemia
kidney biopsy to make diagnosis
treatment of minimal chagne
prednisone - usually dramatic and quick response
treat underlying secondary disease
urokinase plasminogen activating receptor (suPAR)
role in FSGS
Treatment for PRIMARY FSGS
steroids first line
most are steroid resistant
second = calcineurin inhibitors
some targetted thereapy?
recur post transplant!
secondary FSGS
secondary to other kidney disease and obesity
how does primary FSGS present?
NS or asymp prteinuria
normal or elevated BP
how does secondary FSGS present?
NON-nephrotic preinuria, decreased GFR
How do you treat secondary FSGS?
ACEI/ATR blocker
collapsing glomeruloathy etiology
variant of FSGS
characterized by dedifferentiation and proliferation of podocytes with collapse of glomerular tuft
HIV nephropathy (infects podocytes causing proliferation)
or infections, meds, malignanc
treatment of collpsing glomerulopathy
anti retroviral
correct underlying
ACEI/ARBs
APOL1
worse prognosis in FSGS, more likely to develop kidney failure in african americans
1 risk allele = prptection from trypanosomes
2 = risk for kidney failure
histopath of membraous nephropathy
characterized by C3, IgG deposits
SUBEPITHELIAL
how does membranous nephropathy present
NS or asymptomatic proteinuria
outcomes of membranous nephropathy
25% spontaneous remission
50% persistent proteinuria
25% renal failure
treatment for membranous nephropathy
ACEI/ARB
prednisone/calcineuron?
primary membranous nepropathy pathogenesis
IgG antibody to podocyte ag (PLA2R)
Ab fixes compliment and C3 is present in renal tissue
Here would be the podocyte (brownish stuff)
- Expresses the antigen (phospholipase A2 receptor)
- Ab is generated to that autoimmune antibody receptor that binds the receptor
- That then activates complement à destroys the podocyte and gives you this disease
- To remind you, an Ag-Ab complex can activate complement à which ultimately can form this membrane attack complex

PLA2R
phospholipase A2 receptor on the membrane of the podocyte
IgG ab bonds to it and fixes comp
Here would be the podocyte (brownish stuff)
- Expresses the antigen (phospholipase A2 receptor)
- Ab is generated to that autoimmune antibody receptor that binds the receptor
- That then activates complement à destroys the podocyte and gives you this disease
- To remind you, an Ag-Ab complex can activate complement à which ultimately can form this membrane attack complex

secondary membranous nephropathy pantogenesis
trapping of preformed antibody-angigen complexes leading to fixation of complement and podycte damage
SLE
syphilis
malaria
hep B
drugs
tumor
rapidly progressive glomerulonephritis (RPGB)
presentations OF nephritic syndrome that are emergent
based on percent of cresecents (not time!!)
require urgent treatment
post infectious glomerular nephritis
small circulating immune complexes of low-avidity antibody an oligovalent angigen (any infection can cause)
clinical presentation of post infections GN
nephritic syndrome 1-2 wks after strep infection (skin, throat)
pathology of post infectious GN
subepithelial deposition of immune complexes
granular on immunoflorscence
clinical course of post infectious gn
most recover in a couple weeks
control - BP, diuresis, infection
IgA nephropathy pathology
mesangial IC deposits
with IgA and usually C3 and IgG
- Shown biopsy with immunofluorescence
- Slice of kidney à primary Ab against IgA, IgG or IgF à secondary Ab with something that can be detected fluorescently
- In this case see IgA deposited in kidney in mesangium and around glomerular capillaries (L piecture)
- You also see complement (R picture)
- This would be enough to give you a dx of IgA nephropathy

pathogenesis of IgA nephropathy
- incrased levels of galactose deficienct IgA
- production of unique auto antibodies
- formation of pathogenic IgA contianing ICs circulating
- mesangial deposition and glomerular injury
Somehow you get this galactose-deficient IgA à produce unique auto antibody à some systemic (maybe driven by a third factor like infection) à deposition of immune complex à activate immune response à inflammation

membranoproliferative glomerulonephritis causes
hep B, hep C, malignancy, eds
can present w systemic signs of vasculitis and renaly insufficiency and nephritic syndrome
skin rash etc
treat underlying
ANCA associated vasculitis
antibodies to proteins expressed in neutrophil
binding of abs to neutrophil plasma membrane leads to neutrophil activation which causes kidney disease
get autiantibodies by molecular mimicry - present protein that looks like self
treatment of ANCA-associated vasculitis
cancer model
inductions (steroids and abs)
plasmapheresis if severe renal impairment
interfere w immune system
anti-gbm mediated glomerularnephritis
i.e. goodpastures (+ pulmonary hemorrhage)
auto antibodies against alpha3 chain of collagen IV in renal and lung BM
ab activates compliment
presentation of anti-GBM mediated GN
oliguria
advanced renal failure
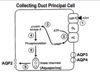
ADH action on collecting duct
prinipal cell

AQP2, 3, and 4 are all affected by ADH. ADH attaches to the V2 receptor on the basolateral surface of principle cells in the collecting duct. That activates GTP-associated protein which activates adenylate cyclase. cAMP activates PKA in the cytoplasm and PKA in the nucleus. In the nucleus, PKA phosphorylates various transcription factors and increases the synthesis of AQP2, 3, and 4. Activation of PKA in the cytoplasm phosphorylates AQP2. AQP2 is located in a subapical region (in vesicles) and when it is phosphorylated, the aquaporin 2 moves to the apical membrane, fuses with it, and makes it water permeable. When AQP2 in the membrane is dephosphorylated, it moves back to the subapical region and vesicles accumulate. There is a shuttling of AQP2 between apical membrane and subapical region that is ultimately under the influence of ADH.
AQP3 and 4 are located on the basolateral surface and are not thought to shuttle between sub-basolateral membrane and membrane; they tend to stay on the membrane. You can see that water flows in the CD across AQP2 on the apical surface and out the basolateral membrane via AQP3 and 4. Exactly how water transport occurs through the cytoplasm is currently unknown.
AQP 2
AQP2 is located in a subapical region (in vesicles) and when it is phosphorylated, the aquaporin 2 moves to the apical membrane, fuses with it, and makes it water permeable. When AQP2 in the membrane is dephosphorylated, it moves back to the subapical region and vesicles accumulate. There is a shuttling of AQP2 between apical membrane and subapical region that is ultimately under the influence of ADH.

AQP
AQP2 is located in a subapical region (in vesicles) and when it is phosphorylated, the aquaporin 2 moves to the apical membrane, fuses with it, and makes it water permeable. When AQP2 in the membrane is dephosphorylated, it moves back to the subapical region and vesicles accumulate. There is a shuttling of AQP2 between apical membrane and subapical region that is ultimately under the influence of ADH.
AQP3 and 4 are located on the basolateral surface and are not thought to shuttle between sub-basolateral membrane and membrane; they tend to stay on the membrane. You can see that water flows in the CD across AQP2 on the apical surface and out the basolateral membrane via AQP3 and 4. Exactly how water transport occurs through the cytoplasm is currently unknown.
what can impair autoregulation?
NSAIDs, ACEi, vascular disase, CKD, chronically elevated BP

pathophysiology of ATN/AKI
- This is a little more detailed view of the various mechanisms that are proposed to explain why GFR falls so much in tubular injury
- Note: the peritubular capillary is shown at left
- If you have shock, toxins, you have endothelial damage, an increase in adhesion molecules, and the inflammatory cells can enter the tubular lumen
- There are cytokines that damage the tubular cells
- In the proximal tubule (to the right of the peritubular capillary), you either lose the integrity of the cells, or lose cells entirely
- When this occurs, there is nothing to protect the basement membrane from allowing the filtrate in the lumen to leak back into the circulation
- You can filter urea, for example, but if it leaks back, it will not be excreted
- These cells lose microvilli, some become necrotic or apoptotic and slough off in the tubular lumen à they no longer function like normal tubular cells
- The image at right shows a region later on in the distal portion of the nephron
- That image is a picture of tubular cells now contacting Tamm-Horsfall proteins, polymerizing with the protein, adhering to one another and forming a cast that now obstructs the tubular lumen and will prevent urine flow
It is a combination of ischemic damage, necrosis, loss of normal polarization of the tubular cells, cast formation, back leak à these all lead to renal failure when you have toxic or ischemic damage to the tubules

alterations in tubule cell structure after ischemic AKI
loss of brush border and polarity
necrosis and apoptosis
sloughing off of cells w lumenal obstruction
dedifferentiation of viable cells

tubuloglomerular feedback
decreased Na reabsorption in the proximal tubule increases delivery to the MD signalling the glomerulus to reduce GFR

timecourse of glomerular hemodynamics in obstruction
- The kidneys are designed to protect themselves
- The first thing that happens in obstruction is the tubular pressure goes up
- The kidneys think they may need more blood flow in order to overcome that
- Afferent resistance will go down
- Glomerular pressure will rise in an attempt to balance tubular pressure
- Depending on how severe the obstruction is, the glomerular filtration rate (GFR) may not initially fall
- If the tubular pressure stays very high, the kidney now knows any further perfusion will deleterious
- Therefore, you have renal vasoconstriction that causes glomerular pressure to be lower and GFR falls still further
- After you relieve the obstruction, it takes some time—not too long, but a little while—for the decrease in tubular pressure to be reflected in a decrease in afferent resistance
- This is less than 24 hours, but you may not see an immediate increase in GFR
- It may take time because the tubular obstruction has set off all of these compensatory mechanisms to try to initially maintain GFR and then prevent further damage
- The take-home message in obstruction: tubular pressure goes up, GFR goes down
- When you relieve the obstruction, this recovers within 24 hours

TGF beta in secondary ckd
endothelial cells: mechanical stretch activate EC to secrete factors that cause fibrosis
mesangial cell - mechanical stretch - narrowing

how to decrease glomerular capillary pressure in CKD?
- control BP
- use ACEI as first line therapy

How do we prevent the progression of CKD
inhibiting RAS

how does proteinuria contribute to progression of ckd?
increased filtered albumin is taken up by PT
accumulate in cytoplasm - perturbation of cell function - recruit macrphages and T cells, activate TGF beta - bind to cells
fibroblast proliferation and matrix depositioon

CKD and acedemia
CKD causes acidemia - increase interstitial fibrosis

This is just to highlight, because it is really important, that a lot of the things that you do, there’s not good data. There is good data for angiotensin system for progression. There is good data if you can decrease proteinuria. There is also data for treating the acidosis, for preventing progression. I mean, controlled trials that if you treat with bicarbonate and get the serum bicarbonate to normal, you will preserve kidney function. So ammonia can increase renal acid production, can activate complement, can activate a cytokine called endothelin.
HIF-1
There is a transcription factor regulated by oxygen called HIF-1 (hypoxia inducible factor), which under normal conditions when oxygen is present, it is degraded. When oxygen is very low, the enzyme that causes it to get degraded, a prolyl hydroxylase, gets inhibited, and so HIF-1 is not degraded. It accumulates, it goes to the nucleus. It turns on erythropoietin, which then circulates and goes to the bone marrow, and commits RBCs to differentiate. If you don’t have kidney’s, your erythropoietin levels are low.

Ca and Vit D
vitamin D needs to be activated by 2 ways, there is a 25 hydroxylase in the liver and then a one (hydroxylase?) in the kidney. So 1,25 (OH)2D is your active vitamin D. If you don’t have kidney function your 1,25 is decreased. That’s important for calcium reabsorption for the gut. So that’s another reason why your calcium could be low.
directly suppresses PTH in parathyroid

tadeoff hypothesis
decreased nephron mass - decrease PO4 clearance - increased PO4 - decrease C, decreased D - increased PTH
also high FGF 23

FGF 23
, fibroblast growth factor 23, this is actually one of the major stimuli for the kidney to decrease reabsorption of phosphorus. And it blocks phosphorus reabsorption by the kidney. So what happens is as you lose kidney function, phosphorus goes up, FGF23 is made to try to… is one of the other… maybe more important than PTH. FGF23 then causes increased phosphate loss, but FGF23 does a lot of other things. It can lower 1,25. It blocks the 1 hydroxylase. So it lowers vitamin D, which has its other effects.

diarrhea acid-base abnormality
hyperchloemic metabolic acidosis
loss of stool (NaCl, KCl, NaHCO3) = gain of H+
decrease serum HCO3, decrease amt filtered
Kidney must reabsorb Na (esp bc high renin, low vol)
Renal Na reapsorption is accompanied by more Cl becaue no HCO3 is available!

lactic acidosis acid-base abnormality
anion gap metabolic acidosis (gain of acid w non-Cl anion)
anaerobic metabolism
H+lactate- added to the plasma
H+ titrates HCO3 - decrease HCO3 - lactate replaces HCO3 in serum

Anion gap calculation
Na - Cl - HCO3 = UA-UC = Anion Gap
nl = 6-12
(major UA is albumin)

Distal RTA (Type I)
distal tubular defect in H+ ion secretion (congenital or acquired)
decreased rate of H+ ion secretion - can’t get rid of daily proton load
urine pH > 5.5 (can’t acidify)
hyperchloremic metabolic acidosis (like gain of HCl)
H+ + Bone –> hypercalciuria and osteoporosis

Type II RTA
proximal tubular defect in H+ ion secretion
impaired HCO3 reabsorption in the PT
congenital or acquired - ocular abnormalities
still able to max acidify urine (pH < 5.5)
hyperchloremic acidosis but patients are in acid balance! To maintain Na - reabsorb Cl (really low HCO3)
No hypercalciuria or stones
if give HCO3 - pee it right out bc can’t reabsorb

contraction alkalosis
loss of fluid relatively high in Cl
leads to a higher [HCO3]
does not account for the kidney!
diuretics cause
