Regulation of Na and H2O excretion week 2 Flashcards
By regulating salt and H2O balance, the kidney is actually controlling ___ and ____ _____.
Why/how do the kidneys have a crucial function in regulating blood pressure?
By regulating salt and H20 balance, the kidney is actually controling osmolarity and blood pressure as well. The amounts of salt and water in the body define blood volume & osmolarity. To achieve salt balance, the kidney must excrete the amount of salt taken in by the body. To achieve H20 balance, the kidney must excrete the amount of H20 taken in by the body. A change in either salt or H20 balance will change the volume and osmolarity body fluids. Since the kidneys ultimately control blood volume, they have a crucial role in regulating blood pressure.
What are the 3 essential elements for generating the medullary osmotic gradient?
The three essential elements for generating the medullary osmotic gradient are:
- The active Na+ reabsorption by the thick ascending limb of loop of Henle.
- The unique arrangement of peritubular capillaries (vasa recta) in medulla.
- The recycling of urea between collecting duct and loop of Henle.
Explain the importance/contribution of active Na+ reabsorption by the thick ascending LOH for generating the medullary osmotic gradient. What would happen if this process was inhibited?
he thick ascending limb reabsorbs solute (i.e. Na+ ) not H2O. This dilutes the tubular fluid. However, it simultaneously concentrates the medullary interstitium (i.e. adding solute without adding an equal amount of water). This is a crucial factor because the medullary osmotic gradient is lost if the Na-K-2Cl symporter of the thick ascending limb is completely blocked and urine will become iso-osmotic (compared to plasma). Portions of the thick ascending limb in the renal cortex also reabsorb more solute than H2O but this part of the cortex does not become hyper-osmotic. The reason is that there are abundant peritubular capillaries and a large blood flow in the cortex that quickly washes out the extra reabsorbed solute.

Explain the importance/contribution of the vasa recta capillaries to medullary osmotic gradient.
The blood flow in the medulla (through the vasa recta) is considerably less than that through the cortex. Low blood flow means that solute can accumulate in the medulla. The low blood flow does not generate the hyperosmotic environment of the medulla but it does help preserve it. Another important aspect is the physical looping arrangement of the vasa recta. Blood moves into the medulla down descending vasa recta and out of the medulla up ascending vasa recta. Although the specifics are yet unclear, solutes and H2O can enter/leave these capillaries (just as in all capillary beds). Thus, the plasma inside the vasa recta osmotically equilibrates with the surrounding interstitium. Plasma moving into the medulla (down the vasa recta) will therefore become more hyper-osmotic. The vasa recta then turn and this hyper-osmotic plasma flows out of the medulla in the opposite direction (up the vasa recta). As it moves up the vasa recta (toward the cortex), its osmolarity returns back to the original value. Imagine if the path of blood flow was straight through the medulla (in at cortex, out at papilla) instead of the hairpin loop arrangement. The solute gained as the blood moves deeper into the medulla would be lost back to the general circulation (not retained in the medulla) and the hyper-osmolarity of the medulla would be quickly washed out (if it could ever develop in the first place). Equilibrating fluids flowing in opposite directions in parallel vessels is an example of a countercurrent exchange system. The main point here is that the low blood flow and looping arrangement of the vasa recta in the medulla are key for preserving the hyperosmotic environment of the medulla.
Explain the importance/contribution of urea recycling for the medullary osmotic gradient.
What 2 solutes contribute to hyperosmolarity of the meduall when the body is conserving H2O?
How may overhydration affect urea recycling?
When the body needs to conserve H2O, the H2O permeability of the collecting duct will be high (up regulated by ADH) and tubular flow may be somewhat low (due to dehydration). If a lot of H2O is reabsorbed from the cortical and outer medullary collecting duct, then the urea concentration in the tubular fluid will rise dramatically (perhaps 50 times higher than it was when it left the loop of Henle). Imagine, the urea concentration in the tubular fluid by the time it reaches the inner medullary collecting duct reaches 500 mOsM. This high urea concentration would provide a substantial driving force for urea reabsorption and urea would move into the inner medulla. Indeed, the urea level in the medulla interstitium may even have time to equilibrate with that in the tubular fluid if tubular flow is slow enough. So….if the tubular urea concentration is 500 mOsM then the urea concentration in the medulla interstitium may approach 500 mOsM as well. Of course, the inner medulla already contains substantial amounts of NaCl and is already hyperosmotic. Thus, the urea added from the inner medullary collecting duct will only increase the hyperosmolarity further. Roughly half the high medullary osmolarity is contributed by urea and half is contributed by NaCl when the body is conserving H2O.
When the body is over hydrated, the H2O permeability of the collecting duct is very low (no ADH present). The tubular flow may be somewhat high (due to the over hydration). The urea concentration in the tubular fluid will not rise substantially before it reaches the inner medullary collecting duct. This is because little (if any) H2O is reabsorbed. There may or may not be a driving force for urea reabsorption form the inner medullar collecting duct. Some urea may be reabsorbed if tubular urea concentration is higher than that in the interstitium. However, urea can actually be secreted into the collecting duct if tubular urea concentration is lower than that in the interstitium. In this latter situation, medullary urea will be washed out and consequently over time medullary osmolarity may be reduced.
Explain this graph.

see slide 6 of notes

State the time frames and mechanisms of control for each of the following:
Short-term BP regultation
Medium-term BP regulation
Long-term BP regulation
slide 7 of course notes

What cells release renin? Where are these cells located?
What 3 signals trigger renin release?
What does renin do? What is the effect on BP?
The kidney does not contribute to short-term BP regulation, but it can reinforce it when a BP challenge is more prolonged. The BP detector here is in the kidney and it senses renal afferent arteriolar pressure. This renal sensing involves granular cells associated with the juxtaglomerular apparatus. These intrarenal baroreceptors are not nerve cells and do not send signals to the brain stem. But, they do receive sympathetic input from the brain stem. When renal afferent arteriolar pressure is low (or in response to sympathetic input), the granular cells release the peptide hormone renin. Renin is a proteolytic enzyme that cleaves angiotensinogen (a peptide produced in the liver) to form angiotensin I. Angiotensin-converting enzyme (ACE) then cleaves angiotensin I to form angiotensin II which is a potent circulating vasoconstrictor. Thus, renin release (triggered by low BP) results in vasoconstriction and increased BP. This response is slower but more persistent than the short-term BP regulation described above. How short-term and medium-term BP regulation combine to control BP after a fast drop in blood volume (possibly resulting from a hemorrhage or even severe diarrhea) is summarized in Figure 5.1. (attached)
Two stimuli that result in renin release (as just described) are changes in renal afferent arteriolar pressure and sympathetic input from the brain stem. There is another intra-renal mechanism that regulates renin release. The macula densa of the juxtaglomerular apparatus detects the amount of NaCl leaving the thick ascending limb of the loop of Henle and then relays this information to the granular cells. The amount of NaCl present in the tubular fluid depends on GFR (which is influenced by BP) and the rate of Na+ reabsortion. When the macula densa senses a low NaCl level, it signals the granular cells to release renin (and visa versa). The macula densa NaCl load sensing (summarized in Figure 5.2) couples medium-term BP regulation with longterm BP regulation through control of renal salt and H20 excretion.

What solutes account for more than 90% of the ECF? How does this relate to controlling blood volume?
Despite the effectiveness of the baroreceptor reflex and the potency of the reninangioensin II sytem, the ultimate factor that determines BP in the long-term is blood volume. Keeping blood volume constant is the job of the kidneys and this takes time. The short-term and medium-term BP regulatory systems may be able to compensate for acute blood volume changes for awhile but it’s the kidneys that are the long-term regulators of blood volume (over hours to days). The kidney does this by adjusting the salt and H20 content of the extracellular fluids. Note that >90% of the osmolarity of extracellular fluid is accounted for by Na+ and the anions (primarily Cl- ) that accompany it. The other ~10% is due to other solutes like K+ , Ca2+, urea, etc. Thus, regulating body fluid osmolarity amounts to regulating Na+ content. Regulating osmolarity amounts to regulating volume. This latter point is not so intuitive so let me quickly explain. The volume of any body compartment (like the plasma) will change as H20 moves in or out of it. Water moves down osmotic gradients so regulating osmolarity is a means of controlling volume. In other words, if the kidneys control Na+ content (and/or the amount of water that Na+ is contained in), then they control volume. If they control volume, then they control BP.
What is diuresis? What is pressure diuresis?
What is natriuresis? What is pressure natriuresis?
What is the mechanism of pressure natriuresis? Where in the nephron does it occur?
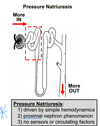
A natriuresis is the excretion of excess Na+ in the urine. Diuresis is excretion of a large volume of urine. Natriuresis resulting from an increase in renal arterial pressure is called a pressure natriuresis. Likewise, a diuresis resulting from an increase in renal arterial pressure is called a pressure diuresis. The mechanism here is thought to be largely hemodynamic without involving any sensors or circulating factors. The concept, however, is deceptively simple. The “simple” part is the underlying concept that if you drive more into the nephron (i.e. due to GFR) then more will come out (i.e. Na+ & H2O excretion). Naturally, this would decrease BP correcting for the increased BP that elevated GFR in the first place. The “deceptive” part here is that the mechanisms are not so clear. For example, there appears to be some unknown (illdefined) mechanism by which increased arterial pressure prompts reduction of apical Na+ transport in the proximal tubule (i.e. promoting Na excretion).
The things you should remember about pressure natriuresis are that:
1) it is driven by physical/hemodynamic factors
2) it does not involve any sensors or circulating factors
3) it is largely a proximal nephron phenomenon. Pressure natriuresis can help in controlling blood volume (and BP) but it is not very useful for independently controlling Na+ and H2O balance.
What hormone mediates the most important control mechansim governing Na+ reabsorption independent of H2O?
aldosterone
Where is aldosterone produced? What are the 2 stimuli for its release?
What is the site of action of aldosterone?
What are the cellular effects of aldosterone?
The most important control mechanism governing Na+ reabsorption (independent of H2O) involves aldosterone, a steroid hormone. Aldosterone increases Na+ reabsorption by the principle cells in the collecting ducts. Aldosterone is not produced by the kidney. Instead, it is produced in the adrenal cortex. The circulating level of angiotensin II controls the production of aldosterone. For example, a decrease in BP triggers a baroreceptor response (i.e. a short-term BP response) which results in the release of renin (i.e. medium-term BP response). The renin released increases circulating angiotensin II levels and this in turn stimulates the adrenal cortex to release aldosterone. The aldosterone then acts on the distal nephron (collecting duct) increasing Na+ reabsorption. The result is greater Na+ retention and consequently increased blood volume which of course tends to increase BP (i.e. long-term BP control).
There are two input signals to the adrenal gland that stimulate adosterone secretion. These are increased plasma K+ levels (this will be described later) and increased plasma levels of angiotensin II (described here). Plasma angiotensin II level is determined by plasma renin concentration. Thus, factors that drive renin secretion (i.e. intrarenal baroreception, macula densa & renal sympathetic inputs) will modulate aldosterone secretion. Reduced mean arterial BP (due to hemorrhage, diarrhea, low- Na+ diet, etc) results in increased adrenal adosterone production (stimulated by the renin-mediated rise in plama angiotensin II levels). Of course, increased mean arterial BP will have the converse action.
Aldosterone, like many other steroid hormones, can freely cross cell membranes and thus simply diffuses into its target cell (i.e. the principle cell). In the cell, it combines with mineralocorticoid receptors in the cytoplasm and then the bound-receptor moves to the nucleus where it promotes gene expression and synthesis of new mRNA. The result is production of new proteins that increase the activity (or number) of, 1) apical Na+ channels and 2) basolateral NaK-ATPase pumps. This promotes increased Na+ reabsorption.
As we learned earlier, most filtered Na+ has already been reabsorbed by the time the tubular fluid reaches the collecting duct. Aldosterone action in the collecting duct is essentially “fine-tuning” Na+ output.
How can aldosterone have a significant impact on Na+ levels if most of the Na+ has already been reabsorbed?
As we learned earlier, most filtered Na+ has already been reabsorbed by the time the tubular fluid reaches the collecting duct. Aldosterone action in the collecting duct is essentially “fine-tuning” Na+ output. Indeed, only 2% of filtered Na+ is actually subject to aldosterone regulation. A person will excrete ~2% of filtered Na+ in the complete absence of aldosterone and excrete virtually no Na+ when a maximal plasma aldosterone concentration is present. Intuitively, 2% seems like a small value but it is not in light of how much Na+ is filtered. Consider the following:
Total Na+ filtered per day = GFR (per day) x PNa = 180 L/day x 145 mmoles/L = 26,100 mmoles/day Aldosterone controls 2% of this or 522 mmoles/day. This represents ~30 grams of table salt (NaCl) which is well more than the average person consumes per day. Indeed, the US RDA (recommended daily allowance) of Na is 2.4 g (or about 1 teaspoon) per day. Thus, the aldosterone control system is easily capable of balancing normal Na+ input and output to keep total body Na+ constant.
What is the stimulus for the release of atrial natriuretic peptide (ANP)? What are the effects of ANP?
Atrial Natriuretic Peptide (ANP): ANP promotes Na+ excretion. The main source of ANP is the heart, specifically the atria. The stimulus for ANP production is distension of the atria (which occurs during volume expansion as in persons on a high-salt diet). In the kidney, ANP has both vascular and renal tubular actions in the kidney. It relaxes the afferent arterial and this increases filtration. It inhibits Na+ reabsorption in the collecting duct. It also inhibits the reninangiotensin-aldostrone system at two levels (renin release & ang-II stimulation of aldostrone production).

Where is ADH released from?
What are the 2 factors that stimulate ADH release?
- posterior pituitary
- cardiovascular barorecptors and hypothalmic osmoreceptors
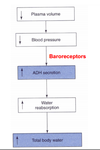
Explain the role of cardiovascular baroreceptors in stimulating ADH release.
What effect may ADH have on blood vessels at very high concentrations?
A decrease in plasma volume (possibly due to diarrhe or hemorrhage) induces increased ADH secretion. The ADH secretion is triggered by neural inputs from baroreceptors (which are located in the carotid sinus, aoric arch and atrium). Decreased BP causes less firing of these baroreceptors and this stimulates the release of ADH into the circulation. Increased BP causes more firing of the baroreceptors and inhibition of ADH release. ADH also has general vasoconstrictor effects when it is present at very high concentrations. A very large decrease in BP (via these baroreceptors) may result in a large increase in circulating ADH. In this situation, ADH may help increase total peripheral resistance which of course will tend to increase BP. At these high concentrations, ADH may also have a similar vasoconstricting action on the renal afferent arterioles and thus reduce GFR (also limiting volume loss to urine).
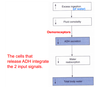
Where is osmolarity sensed? What are the effects of plasma osmolarity on ADH release?
The effect of gaining or losing H20 without corresponding changes in Na+ will result in a change in the osmolarity of the body fluids. Osmolarity is a “sensed” parameter. It is “sensed” by osmoreceptors in the hypothalamus and these receptors communicate with the neurons that release ADH. Increased osmolarity triggers more ADH release. Conversely, decreased osmolarity triggers less ADH release.
Which stimulus for ADH release (baroreceptors or osmoreceptors) usually dominates?
What other factors may effect ADH release?
- Generally, osmolarity usually dominates unless there are very large changes in volume.
- The cells that release ADH also receive inputs from other areas of the brain and thus ADH release can occur in response to things like fear, pain and even inebriation (e.g. alcohol leads to inhibition of ADH release). These more minor inputs should not obscure the fact that the 2 primary control signals for ADH release come from the baroreceptors and osmoreceptors as described above.

What is diabetes insipidus? What are potential mechanisms? What is a sx of this disease?
The importance of ADH is illustrated in patients with diabetes insipidus. This form of diabetes is distinct from diabetes mellitius. The ADH-controlled H20 balance system is disrupted in diabetes insipidus. The problem may be that ADH is not produced (perhaps due to a problem in the brain). Sometimes the principle cells have bad ADH receptors (that don’t “see” the ADH that is present). In this case, the collecting duct’s H20 permeability remains low and unchanging. Regardless of the cause, a clear symptom of diabetes insipidus is a large urine output (i.e. diuresis).
What is free water clearance? Explain how it is calculated.
How can renal H20 handling be assessed? The answer is to determine a quantity that is called free-water clearance (or CH2O). Free-water clearance refers to the volume of solute-free H20 excreted by the kidneys. Determination of CH2O is different than a standard clearance calculation. Renal H20 handling involves separating H20 from solute to generate either dilute or concentrated urine. In performing this separation, the kidneys in a sense generate a volume of H20 that is free of all solute. In other words, dilute urine could be viewed as having two parts. One part that is solute-free H20 and the other part is solute-containing H20. To quantify the “virtual” solute-containing part, osmolar clearance (COSM) is used. The COSM (ml/min) is calculated as follows.
COSM= UOSM x V/POSM
The terms UOSM and POSM are the osmolarity in the urine and plasma, respectively. In words, the COSM is the volume (ml/min) of iso-osmotic fluid that is cleared from the body. Once you know the COSM value, calculating the volume (ml/min) of the “virtual” solute-free H20 portion of the urine (or CH2O) is strait forward. CH2O would logically be the total urine volume (V) minus COSM.
CH2O= V- COSM
What does it mean when CH2O is zero? When it is positive? When it is negative?
When CH2O is zero, it means that no solute-free H20 is excreted and the urine is isoosmotic to the plasma. When CH2O is positive, it means that excess H20 was excreted and the urine is hypo-osmotic to the plasma (i.e. it’s dilute). When CH2O is negative, it means that H20 was retained by the body and the urine is hyper-osmotic to the plasma (i.e. it’s concentrated).

