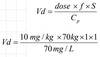Pharmocokinetics and Pharmacodynamics Flashcards
Lecture 1 (Cuddy)
Explain pharmacokinetics
What the body does to the drug. Do I have enough of the drug to achieve the affect (Cp). The study of how drugs are moved through the body and are encompassed in mechanisms of: Absorption Distribution Metabolism Excretion Think Kinetic (movement)
What factors affect absorption of a drug?
- Molecular weight (permeability to cross membrane drops off at >350 grams/mole) 2. Blood flow (gastric blood flow is 150ml/min in stomach vs. 1L/min in sm. intenstine) 3. Solubility (aqueous solutions are absorbed most rapidly followed by oil solutions, suspensions, solid dosage forms) 4. Concentration gradient (higher=more absorption) 5. Disintegration and dissolution 6. Partition coefficient (drug in oil phase/aqueous phase) 7. Transporters (in or out) 8. pH partition theory (Henderson-Hasselbalch) (most are weak acids/bases and where the drug is is a major factor of ionizaiton)
Relate ADME to kinetics and dynamics
After a drug is administered (if orally) it goes through the portal system via the portal vein where it travels to the liver and is either distributed to the plasma (systemic circulation) or metabolized to the kidney where it can either be reabsorbed or eliminated in the urine. Some drugs can expire through the air (alcohol).
What is the definition of distribution and how does it relate to body water?
Protein binding and pH partition as well as total Kg of patient all influence the distribution of the drug. To apply this concept use the loading dose.
What are the non-oral/parenteral routes of drug administration?
- Rectally (think with children, not as popular anymore because better options are available)
- Percutaneously (skin)
- Intravenous (directly to plasma/circulation)
- Intramuscular (absorbed in muscle then to circulation)
- intrathecal (CSF to the brain then plasma)
- Inhalation/pullmonary (Lot of surface area and absorbed to plasma and can be expired through air) ex. albutero/insulin
- Sublingually (no first pass, ex. nitroglycerine - will not work if you swallow it)
Explain the process of oral absorption.
The drug disintegrates in the stomach into smaller particles (dissolution) where it enters into solution and is (absorbed) in the small intestine (site of maximal absorbance/location of large surface area and a lot of blood circulation). There it enters the portal vein where it travels to the liver where it is further metabolized and then travels through blood to site of interest.
Explain the process of oral absorption.
The drug disintegrates in the stomach into smaller particles (dissolution) where it enters into solution and is (absorbed) in the small intestine (site of maximal absorbance/location of large surface area and a lot of blood circulation). There it enters the portal vein where it travels to the liver where it is further metabolized and then travels through blood to site of interest.
Why is the onset of action of a drug important clinically?
If a drug is taken orally, it has to go through the portal system where factors influence how fast (kinetics) the onset will be. Different drugs that a patient might be on (narcotics) might slow down gastric emptying, as well as other factors influence this. If a patient wants to be asleep on a plane ride, you want it to work instantly.
What factors influence gastric emptying?
- Volume of meal 2. Meal composition (high fat slows it down) 3. Viscosity (Increased viscosity reduces rate) 4. Osmotic pressure (Increased pressure increases rate) 5. Position (may be faster when lying on right side) 6. Drugs (Opioids, anticholinergics, ethanol, bile salts and acidification slow emptying. Bicarbonate and metoclopramide (reglan) speed up emptying rate)
What are some examples of topical preparations?
-Creams/ointments -Patches (estrogen, contraceptives, antihypertensive, analgesia, urinary incontinence, antiparkinson, smoking cessation, abtianginal, topical anesthetic) -Slow absorption and no first pass effect, prolonged duration of action
Explain the first-pass effect
a phenomenon of drug metabolism whereby the concentration of a drug is greatly reduced before it reaches the systemic circulation. It is the fraction of lost drug during the process of absorption which is generally related to the liver and gut wall clearance.
What are the main concepts of drug absorption/bioavailability?
Rate and extent!
- The extent of absorption reflects the total amount of drug entering the systemic circulation (it is not time dependent)
- The rate of absorption determines how quickly the drug enters the systemic circulation. The rate typically changes with time.
Bioavailability/How is rate assessed?
By the inspection of Tmax and Cpmax
(rate is a key determinant of peak concentration and peak effect for many drugs)
Bioavailability?
Rate and extent to which the active unchanged ingredient or active moiety is absorbed from a drug product and which becomes systemically available.
Bioavailability/How is extent assessed?
By inspecting the AUC (Area under the curve).
- Affected by absorption AND intestinal wall and first pass clearance in the gut.
- The extent of absorption (F) is calculated by the extent of absorption from the gut (f) and the hepatic extraction ratio (ER).
F= f x (1 - ER)
What is bioequivalence?
Comparable bioavailability between drugs.
The absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study. Assumption is the clearance of the drug is the same.
AUC/Tmax/Cpmax have to be <20% different to be deemed equivalent (FDA)
Equation for absolute bioavailability/what does it tell you?
AUC PO/AUC IV
Different routes/same drug It is telling you that the fraction of drug administered which becomes systemically available. Comparison is made to IV product that F=1.0
Equation for relative bioavailability/what does it tell you?
AUC PO Test/AUC PO
Standard Same active drug/different formulations Fraction of drug which becomes systemically available. Usually oral administration, and same dose.
If you have an F (extent of absorption) that is small, what is that telling you about the first pass clearance?
It means that the drug will be broken down in the stomach and very little of the drug will make it to systemic circulation. Most anesthesia drugs are given IV because of this phenomenon.
What does an F (extent of absorption) of 1 tell you?
That there is 100% bioavailability for the drug when administered OR and IV. Most of the time if a drug is administered IV it means the F is really small and cannot pass through the GI tract and has a high first pass clearance.
What is the apparent volume of distribution?
Apparent volume of distribution (V) links drug concentration to the amount of drug in the body.
Amount in body (Cp) = Volume (L) x Drug Concentration (mg/L)
Why does the apparent volume not always coincide with the anatomical/physiological volume?
Sometimes the V is very high and much greater than the total volume of water in our bodies (~42L) because the drug has diffused to other tissues, is bound to plasma proteins or partitioning into fat or absorption onto bone.
What are some factors that affect drug distribution between body compartments?
- Differences in tissues to difference in blood perfusion 2. Disease states 3. Lipid solubility 4. Extent of drug protein binding (0-100%) 5. Regional differences in pH - ion trapping
What side of the membrane to drugs most likely concentrate?
the side where they are most highly ionized