Oncology Path Flashcards
A 75 year old man goes to his physician. He feels fine but has some fatigue. On exam, his physician palpates his spleen 2 cm below the costal margin and his liver 3 cm below the costal margin. He also has enlarged lymph nodes in his neck and axilla. He orders a routine CBC with a blood smear that reveals the following. What is in the slide below and what might the man’s diagnosis be?

CLL= Small Cell Lymphocytic Lymphoma:
Presentation:
- May be on routine labs
- Seen in elderly
- Male > Female
- Lymphadenopathy
- Splenomegaly/hepatomegaly
- Fatigue
Labs:
- *- Lymphocytosis**
- Cytopenia
- Monoclonal protein
- Hypogammaglobulinemia
- Autoimmune cytopenias
Complications:
- Autoimmune hemolytic anemia (tx: corticosteroids)
- Immune thrombocytopenia (tx: corticosteroids)
- Red cell aplasia (tx: corticosteroids)
- Infections
- Hypogammaglobulinemia
- Transformation to large cell lymphoma (Richter’s transformation)- poor prognosis
Diagnosis:
- Malignant clone (small, mature) with lymphocytes > 5000/uL
- Frequent bone marrow involvement (below)
Immunophenotype/genetics:
- CD5+, CD19+, CD20+/-
- 40% have v-region mutations in H chain gene
Prognosis based on genetics/immuno:
BAD= CD38+, trisomy 12, Bcl-1
Good= abnormal 13q
Combination:
- monoclonal antibody (Rituximab)
- Purine analog (fluarabine, pentostain)
+ Allogenic stem cell transplant (curative)
Prognosis:
- Only curative with stem cell transplant
- Can live many years without transplant

A 55 year old man with a recurrent history of heartburn visits his physician for a physical. He feels healthy and other than heartburn has no chronic health conditions or significant medical history.
On exam, he has palpable lymph node enlargement in his neck and axilla. A lymph node biopsy is performed and the following is found. What is his diagnosis and treatment?

Marginal zone lymphoma:
Often related to chronic infections/inflammatory states
Presentation= Extranodal; May be associated with:
- *- h. pylori**
- Chronic lung infections
- autoimmune disease (thyroid, Sjogren’s)
- 1/3 multifocal
Pathology:
- Tumors derived from cells surrounding germinal centers (B memory cells)
- Larger than small lymphocytes, have more cytoplasm
- Admixed with plasma cells- can be benign (reactive) or derived from tumor cells
Immunophenotype (not specific):
- sig+ (M>G or A)
- IgD- some cig +
- CD19+, CD20+, CD22+, CD79+, CD5-, CD10-, CD23-, CD43-/+, CD11c +/-
Genetics:
- bcl-2 and bcl-1 negative
Treatment:
- *1. Triple antibiotic therapy for H. Pylori
2. Cyclophosamide-based therapy with Rituximab
3. Radiotherapy for early stage disease** - *Prognosis: good; may recur**
- Response to therapy (antibiotics) less likely with deeper invasion, lymph node metastases, or t(11;18) found
Complications: related to organ involved/therapy
A 50 year old woman comes to the doctor’s office for her annual exam. She has no chronic medical conditions and no history of major illnesses. On physical exam she has a palpable lymph node in her axilla. A biopsy of the lymph node is performed and stained for Bcl-2. What is her diagnosis, treatment, and prognosis?

Follicular lymphoma
One of most common indolent lymphomas (22% non-HL)
Presentation: enlarged lymph node; asymptomatic. Common marrow involvement
Pathology:
- Tumor enlarges, fills entire lymph node with neoplastic follicles, obliterates normal architecture
- Recapitulates nodal germinal center arrangement, germinal center cells
- May transform into larger, more aggressive lymphoma
- Involvement: lymph nodes, neoplastic follicles in extranodal soft tissue
- *- Stains for Bcl-2**
Immunophenotype:
- sig+ = IgM+/-, IgD > IgG > IgA
- CD10+ CD25-, CD2-/+, CD42-, CD11c-
- *** Bcl-2**
Genetics:
- t(14;18)(q32;q21): bcl-2 rearrangment–> IgH control–> overexpression of anti-apoptotic Bcl-2
Therapy: **Chemo, rituximab
- ONLY cure is allogeneic stem cell transplant**
- Remission with autologous stem cell transplant
Prognosis: FLIPI score
- Localized disease: 50% 10-year disease free survival; overall 60-70% survival
- Advanced disease: over 10-year median survival
- Elderly patients: watch and wait
Complications: infections, complication of chemo; can have normal life w/o therapy for several years

A 65 year old man is sent to a GI specialist for a colonoscopy following abdominal pain, fever, and nausea and occult blood in his stool. Polypoid lesions are found in his colon and a lymph node is examined after removal. The sample is stained for Cyclin D1. What is his diagnosis based on the sample below, what is his treatment and what is his prognosis?

Mantle Cell Lymphoma
Seen in Adults only (more common in males): frequently involves extranodal spaces
- *Presentation: presents SICK: can be v. aggressive w**ith tumor lysis syndrome; not cured with standard therapy
- Usually seen in advanced disease
- Bone marrow, Waldeyer’s ring (tonsillar) and GI tract involvement
- *- Found in peripheral blood**
Pathology:
- Most aggressive small cell lymphoma;
- Derives from pre-germinal center (antigen-naive) B lymphocytes in zone around germinal center (mantle zone)
- Cells small to medium
- Irregular nuclei (larger= more aggressive)
- Widespread involvement at diagnosis (bone marrow, GI tract)
- Colon involvement–> polypoid lesions (lymphomatous polyposis coli)
Immunophenotype:
- slgM+, IgD+,
- CD 19+. 20+, 22+, 5+, 10-, 23-, 43+, 11c-
- Nuclear Bcl-1+
* Like B-CLL this B-cell tumor expresses a T-cell marker
Genetics:
- t(11;14) involving Bcl-1–> cyclin D1 overexpression (stain)
- Mutation drives cell cycling
Therapy: multiple options:
- *- Chemo, rituximab
- ONLY cure is allogeneic stem cell transplant**
Prognosis:
- Poor without allogeneic transplant
- High M&M with transplant
Complications:
- Infection
- Complications of chemo
- Tumor lysis syndrome
- Rare visceral perforation

A 30 year old woman presents to the Emergency Room after sudden onset of heart palpitations and confusion. Blood work is performed which shows a blood pH of 7.2, elevated K and low Ca. She has no history of arrythmias or diabetes. A CT scan is performed which reveals a large mediastinal mass. A sample of the mass is taken (below). What is her diagnosis and treatment?

Diffuse large B-cell lymphoma
Most common aggressive lymphoma (31% non-HL)
- Variant= mediastinal large cell (younger women with good prognosis)
Presentation:
- Presents sick; aggressive in tumor lysis syndrome at diagnosis
Pathology:
- Arise de novo (most common)
- derive from lower grade tumor (less common)
- Nodal/extra-nodal
- *- Large cells
- Bizarre nuclei, big nucleoli**
- Abnormal levels of Bcl-6
Immuno: slg+, Cig +/-, CD19, 20, 22, 79a, 5+
Genetics:
- Some positive for bcl-2 rearrangement (follicular cell lymphomas)
- some c-myc positive (gene rearrangment)
- bcl-6 rearrangment
Treatment:
Rituximab
Cyclophosphamide
Doxorubicin
Vincristine, prednisone
(R-CHOP)–> nothing better to date
- Autologous HSC transplantation with relapse
Prognosis: based on IPI
Complications: infections, complications of chemo, tumor lysis syndrome
Below: Spleen filled with diffuse large B-cell lymphoma tumors

A 30 year old man who was diagnosed with HIV three years ago presents to the Emergency room with palpitations and confused mental status. Blood panels reveal elevated K, a blood pH of 7.1 and depressed Ca levels. Additionally he has palpable lymph nodes. A biopsy is performed that reveals the following. What is his diagnosis, treatment and prognosis?

Burkitt Lymphoma:
Caused by Epstein Barr Virus: t(8;14)
HIV= risk factor
More common in men
Endemic variant in West Africa with jaw involvement
Non-endemic may have abdominal disease
- Involve kidneys, ovaries, breasts
- 1/3 bone marrow involvement
Presentation:
- Presents sick
- Aggressive tumor lysis (K elevations, Ca depression, acidemia)- Treat tumor lysis with allopurinol, water
Pathology:
- Small, primitive B-cells
- High mitotic rate
- *- Basophilic cytoplasm with lipid vacuoles**
- **Starry sky pattern (=> macrophages ingesting apoptotic debris)
- Stains positive for MYC (due to t(8;14))**
Immuno: slg+, Cig +/-, CD19, 20, 22, 79a, 5+
Genetics:
* t(8;14)(q24;q32)–> MYC, IgH
t(8;22)(q24;q11)–> MYC IgL
t(2;8)(p12;q24)–> IgK, MYC
- *Treatment of Burkitt:
1. **CNS prophylaxis with intrathecal chemo/high dose methotrexate
2nd line:
Rituximab
Cyclophosphamide
Doxorubicin
Vincristine, prednisone
(R-CHOP)
Prognosis: based on IPI
- Children do better than adults
Complications: infections, complications of chemo, tumor lysis syndrome

A 65 year old man presents to his physician after experiencing some strange symptoms and an enlarging painless mass in his neck. He says that he itches all over and that the node in his neck hurts when he drinks alcohol. A biopsy is performed the reveals the following. What is his diagnosis and treatment?

Hodgkin Lymphoma
Presentation:
Bimodal age distribution (v. young and old)
Nodes enlarge over months
- Starts in neck and works its way down body
- B-symptoms (fever, weight loss, night sweats) more common than in non-Hodgkin’s
- Pruritis
- Adenopathy : cervical, axillary, mediastinal
- Nodal pain on alcohol ingestion
- Enlarged mediastinal mass (SVC syndrome, cough- tracheobronchial compression)
- Bone pain (metastatic involvement)
- Marrow depletion with metastases
Pathology:
See “Reed Sternberg” (RS) cell variant:
- Owl’s eye appearance
- Stains positive for CD30- below (80-100%), CD15 (75-85%), BSAP (B-cell specific activating protein, PAX5 gene product- 90% cases)
RS cells seen with polyclonal lymphocytes, eosinophils, neutrophils, plasma cells, fibroblasts, histiocytes
- High number of associated macrophages
* Need biopsy (open) NOT FNAB
Unable to make intact antibodies
Two types:
- Nodular lymphocyte predominant HL
- Classical HL: subtypes:
- Nodular sclerosis
- Mixed cellularity
- Lymphocyte-depleted
- Lymphocyte-rich
Treatment: ABVD:
Doxorubicin (Adriamycin)
Bleomycin
Vinblastin
Dacarbazime
- 90% cure rate with chemo
+ radiation in advanced disease
**EXCEPT NLP: Chemo, rituximab
- ONLY cure is allogeneic stem cell transplant
Prognosis: good
Index for prognosis (each decreases likelihood of remission):
- serum albumin < 4 g/dL
- hemoglobin < 10 g/dL
- male
- age >= 45 years (elderly who receive similar doses of chemo have same outcomes)
- Stage IV disease (Ann Arbor)
- WBC >= 15,000
- Absolute lymphocyte count < 600/mm3 or < 8% total lymphocyte count

A 65 year-old woman presents to her physician complaining of what seems to be a recurrence of her menopause from 7 years ago. She wakes up sweating in the night. Additionally she has lost 15 pounds in the last two months, which she has been trying to do for years. On physical exam she has a couple enlarged lymph nodes in her neck. A biopsy is performed that reveals the following. What is her diagnosis?

Nodular sclerosing Classical HL
70% of cases
Characteristic Mononuclear CD30+ RS-like cells (lacunar cells)
- Mediastinal involvement
- Favorable prognosis
- *Broad bands of fibrosis separating:**
- lymphoplasmacytic reactive cells
- occasional eosinophils
- neutrophils
- classic RS cells
- *Symptoms of HL**:
- B-symptoms (fever, weight loss, night sweats) more common than in non-Hodgkin’s
- Pruritis
- Adenopathy : cervical, axillary, mediastinal
- Nodal pain on alcohol ingestion
- Enlarged mediastinal mass (SVC syndrome, cough- tracheobronchial compression)
- Bone pain (metastatic involvement)
- Marrow depletion with metastases

A 45 year-old HIV+ woman comes to the doctor complaining of pain in her bones. On physical exam she has an enlarged cervical lymph node, which reveals the following on biopsy. What is her diagnosis and what were her risk factors?

Mixed cellularity classical HL
Seen in HIV+ individuals
Reactive cellular infiltrate with:
- Eosinophils
- small lymphocytes
- histiocytes
- abundant RS cells, variants
Resembles NS-HL without fibrosis
- Less mediastinal involvement
- Seen in cervical lymph nodes
- *- EBV+**
Presentation:
- B-symptoms (fever, weight loss, night sweats) more common than in non-Hodgkin’s
- Pruritis
- Adenopathy : cervical, axillary, mediastinal
- Nodal pain on alcohol ingestion
- Enlarged mediastinal mass (SVC syndrome, cough- tracheobronchial compression)
- Bone pain (metastatic involvement)
- Marrow depletion with metastases

An otherwise health 65 year old man is sent to the lab to have a suspicious lymph node biopsied that reveals the following. RS cells are visible in the slide below. What is his diagnosis and treatment?

Nodular lymphocyte predominant HL
Tumor effaces lymph nodes
- Vaguely nodular
Pathology:
- Popcorn cells
- Lobated nuclei
- Lacks CD30 and CD15
- Expresses sig, other B cell markers
- EBV negative
- Skips lymph node groups, does not involve solid organs
- Excellent prognosis
- Uncommon disease
Treatment:
Chemo, rituximab
- ONLY cure is allogeneic stem cell transplant
- different from AVBD treatment for all other HL
A 78 year old woman presents to your clinic for a regular checkup. On exam you notice some brusing on her shins and elbows- she states she has been noticing more frequent bruising and fatigue. You perform a CBC with differential smear and the following is revealed. Flow cytometry reveals a deletion of the 5q chromosome arm in the abnormal cells What is her diagnosis and prognosis based on the pathology below?

Myelodysplastic syndrome: pseudo-Pelger-Huet cells= hyposegmented granulocytic neutrophils seen in MDS
Below: anemia, two populations of RBCs
MDS= clonal hematopoietic stem cell disorders characterized by marrow failure, peripheral cytopenias, dysplastic morphology
- Ineffective hematopoiesis: increased apoptosis of progenitors, limited response to growth factors
- Abnormalities in proliferation, differentiation, apoptosis of precursors and progeny
- *Epidemiology:**
- Men, > 70 years
- *- Sporadic**
- Risk factors: previous chemo (solid tumor < lymphoma), radiation, toxic exposures (pesticides, benzene)
- Genetic syndromes: Diamond-Blackfan, Schwachman-Diamond, Fanconi’s anemia, Dyskeratosis congenita, congenital neutropenia
Path:
Peripheral blood:
- Anemia (normocytic or macrocytic)- acclerated apoptosis of increased progenitor cells
- dual RBC population (may be transfusion related)
- +/- Neutropenia, thrombocytopenia
- Low reticulocyte count
- Dimorphic red cells on histology
- Dysgranulopoiesis (pseudo-Pelger-Huet cells): abnormal granulocyte nucleus, staining, shape
- Dyserythropoiesis (dysplastic erythroid lineage)
- Ringed sideroblasts in RBC precursors (iron accumulation in mitochondria)
- Dysplastic megakaryocytes
- Dacrocytes (teardrop cells), red cell fragments, rouleaux formation, helmet cells
Bone marrow (perform aspirate and biopsy)
- Hypercellular
- Dysplasia (10% cells in lineage show dysplastic features)
- +/- increased blasts (myeloblasts, monoblasts)
Prognosis:
High grade: genetically unstable (mutator)
- Increased risk of transformation to AML (increased blasts–> increased transformation)
- Survival: 6-30 months
- 7q- genotype
- *Low grade (lack mutator phenotype)
- More stable**
- *- Survival: 6-8 years
- 5q- genotype: seen in older women**
- *Treatment:**
1. Hematopoeitic growth factors: - Recombinant erythropoietic stimulating agent (ESA) - Best in patients with low EPO
- G-CSF
- Epigenetic therapy:
- 5-AZA, decitabine - Immunomodulatory drugs:
- Lenalidomide (for 5q deletion, low risk pts) - Immunosuppressive:
- antithymocyte globulin plus cyclosporine
* *5. HSCT= ONLY cure for MDS** - Iron chelation (for iron overload in transfusion-dependent patients)

A 75 year-old man comes to his physician for a 5-year follow-up of treatment for a mild lymphoma. He recieved chemo and went into remission 4 years ago. He presents with worsening fatigue and doesn’t know if it’s just because he’s getting older or his cancer could be recurring. On a CBC with smear, the following anomaly is noted. What is going on and what were his risk factors?

MDS: ringed sideroblasts
Below: dysplastic megakaryocytes
Types of MDS:
- Refractory anemia
* *2. Refractory anemia with ring sideroblasts (RARS)** - Refractory anemia with excess blasts (RAEB)
- CMML
- Refractory anemia with excess blasts in transformation (RAEB-t)
MDS= clonal hematopoietic stem cell disorders characterized by marrow failure, peripheral cytopenias, dysplastic morphology
- Ineffective hematopoiesis: increased apoptosis of progenitors, limited response to growth factors
- Abnormalities in proliferation, differentiation, apoptosis of precursors and progeny
Epidemiology:
- *- Men, > 70 years
- Sporadic
- Risk factors: previous chemo**(solid tumor < lymphoma), radiation, toxic exposures (pesticides, benzene)
- Genetic syndromes: Diamond-Blackfan, Schwachman-Diamond, Fanconi’s anemia, Dyskeratosis congenita, congenital neutropenia
Path:
Peripheral blood:
- Anemia (normocytic or macrocytic)- acclerated apoptosis of increased progenitor cells
- dual RBC population (may be transfusion related)
- +/- Neutropenia, thrombocytopenia
- Low reticulocyte count
- Dimorphic red cells on histology
- Dysgranulopoiesis (pseudo-Pelger-Huet cells): abnormal granulocyte nucleus, staining, shape
- Dyserythropoiesis (dysplastic erythroid lineage)
- Ringed sideroblasts in RBC precursors (iron accumulation in mitochondria)
- Dysplastic megakaryocytes
- Dacrocytes (teardrop cells), red cell fragments, rouleaux formation, helmet cells
Bone marrow (perform aspirate and biopsy)
- Hypercellular
- Dysplasia (10% cells in lineage show dysplastic features)
- +/- increased blasts (myeloblasts, monoblasts)
Prognosis:
High grade: genetically unstable (mutator)
- Increased risk of transformation to AML (increased blasts–> increased transformation)
- Survival: 6-30 months
- 7q- genotype
Low grade (lack mutator phenotype)
- More stable
- Survival: 6-8 years
- 5q- genotype: seen in older women
Treatment:
- Hematopoeitic growth factors:
- Recombinant erythropoietic stimulating agent (ESA) - Best in patients with low EPO
- G-CSF - Epigenetic therapy:
- 5-AZA, decitabine - Immunomodulatory drugs:
- Lenalidomide (for 5q deletion, low risk pts) - Immunosuppressive:
- antithymocyte globulin plus cyclosporine - HSCT= ONLY cure for MDS
- Iron chelation (for iron overload in transfusion-dependent patients)

A 60-year-old man presents to his physician with recent worsening fatigue and dyspnea on exertion. He is worried there might be something wrong with his lungs. A CBC with smear is performed that reveals the following. Flow cytometry indicates an increase in P210 protein and Bcl-2. What tests should be performed next, what is his diagnosis, prognosis and treatment?

- *Chronic myelogenous lymphoma**: Chronic phase peripheral blood smear
- *Below**: bone marrow from CML
- *1. Bone marrow sample
2. Analysis for t(9;22) (Phl chrom)
3. Prognosis:**
MOST COMMON myeloproliferative disease (15-20% leukemias)
- Seen in 5th-6th decade
- more common in males
Bi or Tri-phasic disease;
Expansion in GRANULOCYTE pool
Chronic phase= insiduous, 2-8 years
- Bone marrow:
- Increased granulocytes (WBC precursors)
- Smaller megakaryocytes with hypolobated nuclei= “dwarf”
- decreased erythropoiesis
- elevated myeloid to erythroid ratio
- reticulin fibrosis - Peripheral blood:
- Increased WBCs (leukocytosis)
- Increased thrombocytes (+ abnormal platelets)
* *- Basophilia
- Anemia correcting on treatment**
- Enlarged spleen due to red pulp infiltration by leukemic cells
Genetics:
Philadelphia Chromosome: BCR-ABL1 positive t(9:22)
- 9= abl1
- 22= BCR (breakpoint cluster region)
–> increased P210 protein leads to:
1) increased proliferation (constitutive tyrosine kinase activity)
2) MYC/BCL-2 transcription–> cells protected from apoptosis (MYC/BCL-2)
- Translocation seen in 90-95% CML patients, some ALL (acute lymphoblastic leukemia)
Clonal evolution: 70% of patients in blast phase; relapse after BMT:
- +Ph (duplication of Ph chromosome)
- +8
- isochromosome 17q
Treatment:
Tyrosine Kinase Inhibitor: Imatinib
- Blocks effects of BCR/ABL fusion protein

A 67 year old man presents to your office with recurrent bruising and fatigue. He has a palpable spleen and you decide to perform a CBC with smear which reveals 15% blasts in the peripheral blood (below). What is the diagnosis based on the chronicity of his disease and the smear results?

CML: accelerated phase
MOST COMMON myeloproliferative disease (15-20% leukemias)
- Seen in 5th-6th decade
- more common in males
Bi or Tri-phasic disease;
Expansion in GRANULOCYTE pool
Accelerated phase
- Increased blasts: 10-19% in PB or BM
- Increased PB basophils (>20%)
- *- Thrombocytopenia/thrombocytosis
- Increasing WBC/spleen size**
- Clonal cytogenetic evolution
Genetics:
Philadelphia Chromosome: BCR-ABL1 positive t(9:22)
- 9= abl1
- 22= BCR (breakpoint cluster region)
–> increased P210 protein leads to:
1) increased proliferation (constitutive tyrosine kinase activity)
2) MYC/BCL-2 transcription–> cells protected from apoptosis (MYC/BCL-2)
- Translocation seen in 90-95% CML patients, some ALL (acute lymphoblastic leukemia)
Clonal evolution: 70% of patients in blast phase; relapse after BMT:
- +Ph (duplication of Ph chromosome)
- +8
- isochromosome 17q
A 57 year old presents to the physician after receiving abnormal lab results. A bone marrow biopsy is performed that reveals the following (below). Flow cytometry tests postive for t(9;22) and he is sent to an oncologist for treatment. What is his diagnosis, treatment, and prognosis?

Chronic myelogenous lymphoma: Blast crisis (bone marrow sample
Below: Peripheral blood sample: CML blast crisis
- Diagnosis: CML: blast crisis
- Treatment: Tyrosine Kinase inhibitor (imitinab) and Bone marrow replacement (after remission achieved)
- Prognosis: less than 1 year survival; 70% relapse after bone marrow transplant
MOST COMMON myeloproliferative disease (15-20% leukemias)
- Seen in 5th-6th decade
- more common in males
Bi or Tri-phasic disease;
Expansion in GRANULOCYTE pool
Blast phase= < 1 year survival
- Increased myeolobasts (20%) or extamedullary blast proliferation
- Abnormal platelets
- Blasts= myeloid (70%) or lymphoid (30%)
Genetics:
Philadelphia Chromosome: BCR-ABL1 positive t(9:22)
- 9= abl1
- 22= BCR (breakpoint cluster region)
–> increased P210 protein leads to:
1) increased proliferation (constitutive tyrosine kinase activity)
2) MYC/BCL-2 transcription–> cells protected from apoptosis (MYC/BCL-2)
- Translocation seen in 90-95% CML patients, some ALL (acute lymphoblastic leukemia)
- *Clonal evolution: 70% of patients in blast phase; relapse after BMT:**
- +Ph (duplication of Ph chromosome)
- +8
- isochromosome 17q
Treatment:
Tyrosine Kinase Inhibitor: Imatinib
- Blocks effects of BCR/ABL fusion protein
- Bone marrow transplant

A 50 year old woman presents to her physician due to an abnormal CBC from two weeks earlier that showed elevated hematocrit (18 g/dL). On history, she reports recent headaches and difficult hearing as well as shortness of breath while working out, but she thought these were due to lack of sleep and aging. Flow cytometry of her blood reveals a JAK2 mutation and bone marrow biopsy is performed that reveals the following. What is her diagnosis based on her symptoms and the pathology below? What is her treatment and prognosis?

Polycythemia vera (bone marrow biopsy)
Diagnostic Criteria:
- Major:
1. Elevated RBC mass (> 25% above mean) or Hb > 18.5 in men, > 16.5 in women
2. **Presence of JAK2 V617F (or similar mutation)** - Minor:
1. Bone marrow: hypercellular (pan-myelosis)
2. Serum EPO below reference range
3. Erythroid colony formation in vitro (endogenous)
Clinical manifestations:
Blood hyperviscosity (due to increased RBCs) causing:
- Headaches
- blurry vision
- Altered hearing
- Mucous membrane bleeding
- Shortness of breath
- Malaise
Splenomegaly
Thrombosis (arterial most common)
- can be in unusual sites (mesenteric, Budd-Chiari)
Pruritis (provoked by warm water)
Vasomotor symptoms (paresthesias)
Path:
JAK2 mutation
- Changes interaction between EPO receptor and JAK2
- Mutation in position 617–> constituitively active JAK–> clonal expansion
- *Bone marrow:
- Increased cellularity**
- Panmyelosis with full maturation (no increase in blasts)
- Mild/moderate increase in WBCs, platelets
- *- No iron stain (all being used in RBC production)
- Increased reticulin fibers**
Peripheral blood:
- Persistant leukocytosis (elevated WBC)
- Persistant thrombocytosis (elevated platelets)
Polycythemic phase= most patients
- 10% go to “spent” phase (anemia)
- 10% show myelofibrosis (with splenogmealy due to extramedullary hematopoieses
- 5-10% transform to AML
Prognosis: Mean survival= 13 years
Terminal events: see cytogenetic abnormalities (trisomy 18, deletion 20q)
- Myelodysplastic transformation
- Leukemic transformation
- Postpolycythemic myelofibrosis
Treatment:
Main objective=
1. prevent thrombosis in high risk patients (> 60 years, history of thrombosis)
2. Alleviate non-life-threatening symptoms:
- microvascular disturbance (headaches, acral parasthesia, erythromelalgia),
- pruritis (responds to JAK-inhibitor)
- symptomatic splenomegaly (hydroxyurea)
* Pruritis related to JAK-STAT signalling-related cytokines
3. Phlebotomy in all patients (target Hct < 50%–> 45% ideal)
A 35 year old woman presents to her physician for an annual physical. On exam her physician notices that her spleen is palpable 3 cm below the costal margin. She orders a CBC with smear which comes back revealing elevated platelets (500,000) in her peripheral blood. Bone marrow biopsy and flow cytometry are performed, revealing a MPL marker on the megakaryocytes. What is her diagnosis based on the biopsy, her labs, and what is the significance of MPL+?

Essential thrombocythemia: bone marrow biopsy (enlarged nuclei visible below)
- *Megakaryocyte clonal, autonomous proliferation**
- Must distinguish from inflammatory/malignant processes
Epidemiology:
- 1/100,000 individuals
- 55 years (M=W)
- Second peak in women ~30 years
- Usually incidental finding
- *Clinical presentation:**
- may see life-threatening bleeding (common in GI tract)
- Erythromelagia (dramatic vasomotor symptoms)= warmth, pain in distal extremities
- Splenomegaly
- Large vessel thrombosis
- May progress to fibrotic phase (like PMF/AML)- rare
- Venous thrombosis to unusual sites or PE
Genetics:
50% due to JAK-2 V617F mutation or MPL mutations
Path:
Proliferation of marrow megakaryocytes
Peripheral:
- Increased circulating platelets (abnormal morphology)
- Normal Hb, WBC
- Splenomegaly
Bone marrow:
- Increased in megakaryocytes
- Abnormal clustering of megakaryocytes
- Enlarged with hyperlobulated (stag-horn) nuclei, abundant cytoplasm
- *Diagnostic criteria**:
- *All** required for diagnosis:
1. Sustained platelet >= 450,000/uL
2. Megakaryocytic hyperplasia (enlarged and mature) in marrow
3. Exclude other myeloproliferative disorders: - CML (no BCR/ABL fusion)
- no PC
- no myelodysplasia
- no PMF (no collagen/reticulin fibrosis)
4. JAK2 V617F mutation or MPL (EPO receptor) mutation, or exclusion of reactive (secondary) thrombocytosis
Treatment:
JAK-inhibitor
Phlebotomy
Aspirin for clotting

A 64 year old woman comes to her doctor for treatment of fatigue and dyspnea. CBC with smear reveals the following abnormality in her RBCs. Her physician refers her to the hospital where they perform a bone marrow biopsy that reveals reticulin deposition in the marrow. Flow cytometry tests negative for t(9;22) and JAK2 mutations. What is her possible diagnosis, prognosis and treatment?

- *PMF: pre-fibrotic stage**: peripheral blood smear revealing dacrocytes (teardrop shaped RBCs)
- *Below:** pre-fibrotic changes in bone marrow
- Survival= 3-5 year median survival (better prognosis due to pre-fibrotic changes
- No treatment besides BMT
- *Proliferation of Megakaryocytes and Granulocyte elements in bone marrow–> reactive fibrosis**
- Fibrosis= response to growth factors produced by megakaryocytes (clonally abnormal hematopoietic cells)
1. Bone marrow: See reticulin fibers
2. Later: overt collagen fibrosis
Marrow fibrosis–> extramedullary hematopoiesis (spleen, liver, etc.)
Epidemiology:
1/100,000
Most common in 6th-7th decade
60% have cytogenetic abnormality
- Unknown cause (could be radiation- seen in Hiroshima survivors, benzene)
- *Symptoms**:
- Anemia (due to ineffective hematopoeisis, hypersplenism)
- Marked splenomegaly (extramedullary hematopoeisis- also seen in liver)
- Fatigue, weight loss, night sweats, fever
- Peripheral edema, early satiety
- Portal HTN (varices, ascites)
- Bleeding and thrombotic events
Diagnostic criteria:
Major (all 3 needed):
1. Collagen fibrosis/prefibrotic disease in marrow
2. Rule out: CML (no BCR/ABL), PV, other MDS
3. JAK2 V617F mutation or other clonal marker (MPL)
Minor (2+ needed):
- Leukoerythroblastosis (low RBCs, WBCs)
- Increased LDH
- Anemia
- Splenomegaly
Progression:
Early:
- Hypercellular marrow
- Prominent, abnL megakaryocytes
- Increased reticulin
Later:
- Increased fibrosis (collagen)
- Reduced hematopoeitic elements
- End stage: osteosclerosis (thickened bony trabeculae–> decreased marrow space)
* Fibrosis in marrow also seen in: PV, CML
Peripheral blood:
Leukoerythroblastosis= increased immature granulocytes and nucleated RBCs (normoblasts) due to:
- Extramedullary hematopoeisis
- Disruption of normal bone marrow-blood barrier (fibrosis)
* Can also be seen in metastatic solid tumors
Marrow fibrosis leads to:
- myelophthisic anemia (weird RBCs made in other parts of body)
- anisopoikilocytosis (abnL RBC shapes + sizes)
- *- Dacrocytes (teardrop-shaped cells)**
- Giant platelets
- Megakaryocytes in circulation
Spleen:
- Red pulp expansion
- Extramedullary hematopoeisis
- Focal splenic infarcts
Prognosis:
Median survival= 3-5 years
Poor prognosis:
- age > 70
- Hg < 10g/dL
- Leukocyte > 25 x 10^9
- Circulating blasts > 1%
- Constitutional symtoms
Plus:
- Platelet < 100,000/uL
- Immature WBCs in peripheral blood
- Abnormal karyotypes
Treatment:
Lack of drug therapy:
Bone Marrow Transplant in patients with median survival < 5 years and leukemia risk > 20%

A 72 year old man presents to his physician for a physical. He reports increasing fatigue and weakness. On physical exam several bruises are noted on his arms and shins and his spleen is palpable 3 cm below the costal margin. A CBC with smear reveals pancytopenia and immature cells (leukoerythroblastosis). A bone marrow biopsy is performed that reveals the following. What is his diagnosis and the criteria that are met in this man’s presentation?

Primary Myeloid Fibrosis: Fibrotic stage
Below: spleen showing extra-medullary hematopoeisis
Proliferation of Megakaryocytes and Granulocyte elements in bone marrow–> reactive fibrosis
- Fibrosis= response to growth factors produced by megakaryocytes (clonally abnormal hematopoietic cells)
1. Bone marrow: See reticulin fibers
2. Later: overt collagen fibrosis
Marrow fibrosis–> extramedullary hematopoiesis (spleen, liver, etc.)
Epidemiology:
1/100,000
Most common in 6th-7th decade
60% have cytogenetic abnormality
- Unknown cause (could be radiation- seen in Hiroshima survivors, benzene)
Symptoms:
- Anemia (due to ineffective hematopoeisis, hypersplenism)
- *- Marked splenomegaly (extramedullary hematopoeisis- also seen in liver)**
- *- Fatigue,** weight loss, night sweats, fever
- Peripheral edema, early satiety
- Portal HTN (varices, ascites)
- Bleeding and thrombotic events
Diagnostic criteria:
Major (all 3 needed):
1. Collagen fibrosis/prefibrotic disease in marrow
2. Rule out: CML (no BCR/ABL), PV, other MDS
3. JAK2 V617F mutation or other clonal marker (MPL)
Minor (2+ needed):
- Leukoerythroblastosis (low RBCs, WBCs)
- Increased LDH
* *3. Anemia**
* *4. Splenomegaly**
Progression:
Early:
- Hypercellular marrow
- Prominent, abnL megakaryocytes
- Increased reticulin
Later:
- Increased fibrosis (collagen)
- Reduced hematopoeitic elements
- End stage: osteosclerosis (thickened bony trabeculae–> decreased marrow space)
* Fibrosis in marrow also seen in: PV, CML
Peripheral blood:
Leukoerythroblastosis= increased immature granulocytes and nucleated RBCs (normoblasts) due to:
- Extramedullary hematopoeisis
- Disruption of normal bone marrow-blood barrier (fibrosis)
* Can also be seen in metastatic solid tumors
Marrow fibrosis leads to:
- myelophthisic anemia (weird RBCs made in other parts of body)
- anisopoikilocytosis (abnL RBC shapes + sizes)
- Dacrocytes (teardrop-shaped cells)
- Giant platelets
- Megakaryocytes in circulation
- *Spleen:
- Red pulp expansion
- Extramedullary hematopoeisis
- Focal splenic infarcts**
Prognosis:
Median survival= 3-5 years
Poor prognosis:
- age > 70
- Hg < 10g/dL
- Leukocyte > 25 x 10^9
- Circulating blasts > 1%
- Constitutional symtoms
Plus:
- Platelet < 100,000/uL
- Immature WBCs in peripheral blood
- Abnormal karyotypes
Treatment:
Lack of drug therapy:
Bone Marrow Transplant in patients with median survival < 5 years and leukemia risk > 20%

A 65 year old man presents to his physician with increasing fatigue, dyspnea, and a new sensation of “pain in my bones”. The physician begins a physical exam and notices enlargement of both the liver and spleen below the costal margin as well as some bumps on his scalp (below). What is the physician concerned that his patient might have and how should he screen for it?

Acute Leukemia:
20% or more blasts in bone marrow
Blasts= immature hematopoietic cells of myeloid or lymphoid lineage
- Monocytic leukemias= increased tissue infiltration, tumor lysis syndrome, bad cytogenetics
Clonal disorder (somatic mutation in hemoatopoietic precursor) of bone marrow–>
1) Accumulation of clonal abnormal blasts
2) Impaired production of normal blood cells
- leads to anemia, infection, bleeding
Clinical presentation:
- Anemia
- Thrombocytopenia
- White blood count can be low or high
- Neutropenia
- Marrow expansion – bone pain
- Leukostasis – high WBC modified by cell size and plasticity (Lung and CNS)
- Tissue Infiltration; Granulocytic sarcoma
(Gums, skin, testes, meninges, retina)
- Organomegaly: Hepatosplenomegaly
- Mediastinal mass or adenopathy - ALL
- Tumor lysis syndrome: uric acid, LDH
- Coagulopathy – especially in t(15;17) or with infection
- *Diagnostics**:
- CBC with smear
- Bone marrow biopsy
- Flow cytometry
Two types:
1) Acute myelogenous leukemia (AML)
2) Acute lymphoblastic leukemia (ALL)
Causes:
- Clonal hematopoietic disorders: CML, PMF, ET, PV, MDS, PNH
- Ionizing radiation
- Oncogenic viruses: HTLV-1 (T-cell leukemia), EBV (mature B-cell ALL)
- Benzene
- Prior chemo with alkylating agents–> myelodysplastic syndromes 4-6 years later (chromosomes 5, 7, 8 abnormalities)
- Prior chemo with Topoisomerase Inhibitors: Epipodophyllotoxin (teniposide, etoposide): 1-2 years later, no myelodysplasia–> progresses right to monocytic leukemia (chrom 11 q23 or chrom 21 q22)
Genetics:
- 20% chance in identical twin with affected twin
- Trisomy 21
- Trisomy 13 (Patau)
- XXY (Klinefelter)
- Bloom’s syndrome
- Fanconi’s anemia
- Ataxia-telangiectasia
A 73 year old woman is admitted to the hospital with increasing difficulty breathing. On physical exam she has noted hepatosplenomegaly. Her daughter states she has been getting tired more often and she recently had two bouts of pneumonia and a skin infection. Her CBC smear reveals the following. What is the most likely diagnosis based on her age, presentation, and the slide below?

- *Acute myeloblastic leukemia**: peripheral blood smear
- Can see large population of blast cells within blood (>20%) with granulocytic features (myeloid origin)
Epidemiology:
Neonates= predominant leukemia form
Childhood/adolescence= Less common than ALL
Adults= 80% of leukemias (incidence increases with age, vs ALL)
See symptoms due to:
1) Expansion of malignant clone (w/ or w/o organ involvement)
2) Deficiencies in normal blood cells
Pathophysiology
1. Leukocytes: low, normal, high
High Leukocyte levels–> alterations in blood flow in organs/compromise function (cells sticker, larger, stiffer):
- Respiratory failure
- Stroke, cerebral ischemia
- Infiltration different organs
- Skin, CNS, gums
2. Organomegaly with other myeloproliferative disorders
3. Platelet deficiency (thrombocytopenia)–> bleeding, bruising
4. Low granulocytes–> infections
5. Suppression of RBCs–> severe anemia, cardiopulmonary symptoms
* See DIC in acute promyelocytic anemia (APL)
* Elderly at increased risk for pancytopenia
Diagnosis:
Labs:
- Decreased hemoglobin, platelets
- Stains for MPO, non-specific esterases
*AUER RODS= condensed granules
- Seen in any AML, more numerous in neoplastic promyelocytes (APL)
- Stain red with Wright-Geimsa
- NEVER seen in normal myeloid/lymphoblastic precursors
Treatment:
CANNOT be cured without HSCT
- Admission: cultures, antibiotics if febrile
- Fluids, allopurinol to treat/prevent tumor lysis syndrome
- M0-M7 treated the same EXCEPT for M3
- Induction therapy: achieve hematological remission (peripheral blood normal)
- Anthracycline for 3 days
- Cytarabine for 7 days
ex: APL= t(15;17)–> retinoic acid + chemo + arsenic
- Retinoic acid induces maturation of tumor cells, prevents accumulation of granules that can cause DIC - Consolidation therapy:
- High dose Cytarabine: particularly good in t(8;21) and inv16- use high dose ARA-C
- Autologous HSCT: after remission, store bone marrow stem cells–> myeloablative chemo–> rescue hematopoeisis with stem cells
- Allogeneic HSCT: replenish cells post chemo, allow for graft-versus-leukemia effect (less likely to be effective in older patients) - Remission induction:
- Anthracycline, cytarabine - Maintenance therapy: APL ONLY
Side efects:
- Effects on proliferating cells (skin, gut, marrow)
- Cytopenias get worse before they get better
- Mucositis
- Infections and bleeding worsen - Anthracycline cardiac effects
A 30 year old man is admitted to the hospital with recurrent pneumonia infections. He has several bruises noted on his arms and legs. A peripheral blood smear reveals elevated neutrophils with all stages present. Based on his age, presentation, and blood smear, what might his diagnosis be? How is this confirmed and treated?

- *AML with t(8;21)**:
- *- Maturation along neutrophil lineage (M2)**
- Occurs predominantly in younger adults
- *- Good prognosis**
- Therapy with high dose ARA-C may be curative.
- AML with t(8;21) and inv(16) cause alterations to proteins which are part of the Core Binding Factor transcription complex.
Pathophysiology
1. Leukocytes: low, normal, high
High Leukocyte levels–> alterations in blood flow in organs/compromise function (cells sticker, larger, stiffer):
- Respiratory failure
- Stroke, cerebral ischemia
- Infiltration different organs
- Skin, CNS, gums
2. Organomegaly with other myeloproliferative disorders
3. Platelet deficiency (thrombocytopenia)–> bleeding, bruising
4. Low granulocytes–> infections
5. Suppression of RBCs–> severe anemia, cardiopulmonary symptoms
* See DIC in acute promyelocytic anemia (APL)
* Elderly at increased risk for pancytopenia
Diagnosis:
- Decreased hemoglobin, platelets
- Stains for MPO, non-specific esterases
*AUER RODS= condensed granules
- Seen in any AML, more numerous in neoplastic promyelocytes (APL)
- Stain red with Wright-Geimsa
- NEVER seen in normal myeloid/lymphoblastic precursors
Immunophenotype:
- Stem cell marker (CD34) in myeloid or lymphoid basts
- Granulocytic antigens: CD117, CD13, CD15, CD33, MPO
- Monocytic antigens: CD14, CD64
- Erythroid marker: glycophorin A
- Megakaryocytic antigens: CD41, CD61
* TdT not usually seen in AML, usually in ALL
A 32 year old woman comes to her physician complaining of a new onset illness. She is febrile and tachypnic and tachycardic. A blood smear is performed that reveals the following (abnormal eosinophils, other abnormal blast cells). Based on her age, the type of cells present, and her symptoms, what is her condition and treatment?

AML with inv(16):
Usually shows monocytic and granulocytic differentiation (myelomonocytic)
Characterized by conspicuous presence of abnormal eosinophils (M4Eo)
Occurs predominantly in younger adults
Good prognosis
Therapy with high dose ARA-C may be curative.
AML with t(8;21) and inv(16) cause alterations to proteins which are part of the Core Binding Factor transcription complex.
A 65 year old woman presents to the emergency room with fever, weakness and diffuse patchy bruises (petechiae) on her extremities. Her husband said she wasn’t feeling well the past couple of days but she was really not well this morning. She is started on IV antibiotics and fluids, and a D-dimer test is run (due to the petechiae) which is positive. It is determined she is in DIC. Based on the blood smear below, what is her most likely diagnosis, treatment, and prognosis?

AML t(15;17)= acute promyelocytic leukemia (APML)
Proliferation of abnormal promyelocytes with Auer rods
t(15;17)(22; q11.2-12) –> PML–RARa fusion protein –> block in differentiation of promyelocytes
- Unique immunophenotype
- Clinically associated with DIC
- Typically occurs in adults of middle age
- Sensitivity to ATRA and Arsenic
- Good prognosis when optimally treated
- *Treatment**:
1. Trans Retinoic Acid (ATRA) targets the molecular defect- pushes cells through differentiation - Retinoic acid syndrome as cells differentiate (fever, pulmonary infiltrates, hypoxia)
- Combined with anthracyclines (idarubicin, daunorubicin)
- Maintenance therapy important unlike other AML subsets
- *- High percentage of cures**
- Coagulopathy may get worse before it gets better
2. Arsenic Trioxide - Also induces cellular differentiation
- Can produce “Retinoic acid syndrome” (aka differentiation syndrome)
- Decreased role for standard chemotherapy when combined with ATRA
A 73 year old man presents to the Emergency room with sudden onset fever, chills, and diffuse pain he says is “in my bones”. On physical exam he is tachypnic, tachycardic, febrile, and normotensive. Additionally he has a palpable enlargement of several lymph nodes in his neck and axilla. He is put on IV antibiotics and fluids and a blood draw is performed that reveals the following when stained with TdT. What is his diagnosis, and based on the presence of the protein P190, what are the genetics of his disease, treatment, and prognosis?

ALL- TdT staining
- t(9;22)= BAD prognosis (25% adult AML, 3% child AML)–> 190kD fusion protein
12% of all Leukemias
60% of leukemias for people < 20 years
MOST COMMON malignancy in patients < 15 years (25% of all malignancies, 75% of all leukemias)
- Bimodal distribution: peaks at ages 2-5, again in 60s
Subdivided into B-cell and T-cell
Clinical manifestations:
Manifestations similar to AML
May also involve:
- CNS/meninges
- Bone marrow necrosis: pain, fever, high LDH levels
- Painless testicular enlargement
- Mediastinal mass or adenopathy
- No symptoms (v. rare)
Labs:
Diagnose with peripheral blood, bone marrow morphology, flow cytometry, cytogenetics, molecular genetics
- *Peripheral blood (below**)
- Decreased hemoglobin, platelets
- May or may not see leukemic cells
Bone Marrow:
- replaced by malignant clone
Immunophenotype:
- CD10+, CD19+, TdT, intracytoplasmic IgM+, CD22+, CD79a+, HLA-DR+
treatment:
ALL in children= curable
- NOT in neonates, infants
- After 12, cure rate decreases with age
- *1. Induction therapy:**
- Glucocorticoids, vincristine, L-asparaginase
- CNS prophylaxis
- *2. Intensification consolidation therapy:**
- Antimetabolite agents (methotrexate, 6-mercaptopurine)
- *3. Maintenance Therapy:**
- Required for children with ALL (other than mature B-ALL): methotrexate, 6MP
- Unclear value for adults
- *4. Allogeneic HSCT:**
- Children with high risk cases
- Less successful in adults
- *- Standard of care in Phl ALL t(9;21)**
- *5. Tyrosine Kinase Inhibitor (Imatinib)**
- treatment for Phl ALL
Prognosis:
Good prognosis:
- Age > 1, < 12
- Hyperploidy > 50 chromosomes/cell
- ETV6-CBFA2 fusion
- t(12;21)
- *Poor prognosis:**
- *- Phl chrom (esp in adults)**
- CNS disease
- Age < 1, > 12
- Rearrangement of MLL gene (Chromosome 11q23)
- High WBC
- co-morbidities

A 6 year old boy was brought to his pediatrician because of increasing fatigue and bruising. His mother was concerned he may have anemia based on what she read online. On physical exam the boy appears pale and has diffuse brusing on his arms and legs. He is not distressed. The physician performs a blood draw for a CBC with smear that reveals the result below. A chromosomal analysis reveals 58 chromosomes in the abnormal cells. What is his probably diagnosis, treatment and prognosis based on his age and presentation?

- *B-ALL**= most common form of ALL (80-85% of ALL)
- More favorable prognosis in Children
- More common in children < 12 years or adults > 60 years
Types of B-ALL:
- MLL= 11q23 rearrangment
- Bad prognosis
- may occur in utero
- seen in v. young infants (< 1 years)
- High WBC count
- CNS involvement at diagnosis
* *2. B-ALL with hyperdiploidy= > 50, < 66 chromosomes, no structural abnormalities
- Seen in children (rare in adults/infants)
- good prognosis** - B-ALL with t(9;22)= Bcr-ABL1–> Philadelphia chromosome mutation
- Seen more commonly in adults (25% of ALL) vs 3% in children
- Prognosis poor; improved outcomes with tyrosine kinase inhibitors
- *Therapy**:
- determined by age of patient
- Combination + corticosteroids
- “sanctuary”= prophylaxis of CNS
- Transplant high risk cases
- Risk for Tumor lysis syndrome: allopurinol + fluids
- *1. Induction:**
- Glucocorticoids, L asparaginase, vincristine
- CNS testing, prophylaxis
- *2. Intensification/consolidation:**
- Shortly after induction phase recovery:
- Antimetabolites (methotrexate, 6-MP)
- CNS prophylaxis, treatment
- *3. Maintenance therapy:**
- Methotrexate, 6MP for children
4. Allogeneic HSCT: - Children with high risk cases
- Less successful in adults
- Standard of care in Phl ALL t(9;21)
5. Tyrosine kinase inhibitor (Imatinib) - *- Phl ALL t(9;21)**
Esophagus specimen

Squamous carcinoma of esophagus:
2-3 cm above GEJ
- Upper 2/3 of esophagus
- Spread to cervical/paratracheal nodes
Risk factors:
Squamous cancer:
1. Smoking (5-10-fold risk)
2. Alcohol (+ Smoking)
3. HPV
4. Chronic esophagitis
Esophageal specimen

Adenocarcinoma of esphagus:
Arises in lower 1/3
Often from Barrett esophagus
- Inactivation of multiple tumor suppressor genes
Metastasizes to intraabdominal nodes
Risk factors:
- Barrett’s esophagus
- Obesity, male, heartburn, older age, white
- H. pylori
- Smoking
- High salt diet
- Inherited syndromes (1-3%): diffuse, e-cadherin mutations, CDH1
Diagnosis:
UES + biopsy
- Stain specimen for HER2 (in 20% of cancer)
PET/CT

Submucosal layer of stomach

Stromal tumor of stomach
Smooth muscle tumor: GIST
- Occur anywhere in GI tract
- Spindle cells
- Usually benign (Gastric)- intestinal more aggressive
- Immuno+ for c-kit (CD117)- treat with Imatinib
- Submucosal
Sample from lining of stomach

Hyperplastic polyp:
Tortuous hyperplastic glands, slight inflammation
- Dysplastic glands, background of intestinal metaplasia seen
- At risk for malignant change
- Adenomatous polyps and FAP–> ACA (adenocarcinoma)
Sample from gastric lining

Gastric tubular adenoma
Tortuous hyperplastic glands, slight inflammation
- Dysplastic glands, background of intestinal metaplasia seen
- At risk for malignant change
- Adenomatous polyps and FAP–> ACA (adenocarcinoma)
Gastric lining

- *Early gastric cancer**:
- Limited to submucosa, mucosa
- Has access to vasculature and lymphatics
- Can see inflammation subadjacent to tumor
Type I: protrudes into lumen
Type II: Superficial, flat lesion
Type III: malignant ulcer

Advanced gastric cancer- polypoid
Invades into muscularis, lamina propria
- *1. Polypoid: protruding mass**
2. Ulcerating
3. Diffuse infiltrating: - Creates fibrous stomach, highly aggressive
- “linitis plastica”– leather bottle
- Tumor cells invade deeply, inocuously- can look like inflammatory infiltrate
- Stain for mucin (positive)
- Signet-ring like appearance to cells
*50% have e-cadherin mutations
- Signalling molecule for proliferation
*Most gastric cancers found in advanced stage (western world)
Sample from stomach

Advanced gastric cancer: ulcerating
Invades into muscularis, lamina propria
- Polypoid: protruding mass
* *2. Ulcerating** - Diffuse infiltrating:
- Creates fibrous stomach, highly aggressive
- “linitis plastica”– leather bottle
- Tumor cells invade deeply, inocuously- can look like inflammatory infiltrate
- Stain for mucin (positive)
- Signet-ring like appearance to cells
*50% have e-cadherin mutations
- Signalling molecule for proliferation
*Most gastric cancers found in advanced stage (western world)
Sample from stomach

Advanced gastric cancer- diffuse infiltrating:
Invades into muscularis, lamina propria
- Polypoid: protruding mass
- Ulcerating
* *3. Diffuse infiltrating:**
- Creates fibrous stomach, highly aggressive
- “linitis plastica”– leather bottle
- Tumor cells invade deeply, inocuously- can look like inflammatory infiltrate
* *- Stain for mucin (positive)**
- Signet-ring like appearance to cells
*50% have e-cadherin mutations
- Signalling molecule for proliferation
*Most gastric cancers found in advanced stage (western world)
Early refers to size, not duration; early lesions may have spread to nodes
- No necessarily precursors
- Even with metastases, more curable
- “early” gastric cancer= misnomer, as it has different biological morphology

Lining of stomach: secretes serotonin

NETs: carcinoid
GI tract NETs= low grade cancers
- Often polypoid, may ulcerate
- Small round regular cells form nests, cords, gland-like structures
Stomach: high circulating levels of gastrin, gastritis
Appendix: almost always benign
Small intestine: most aggressive
Histology= not predictor of malignancy
Predictors of malignancy:
- Endocrine functionality= produces serotonin (diagnostic test for 5-HIAA urine levels)
- size + invasiveness
- *Can cause carcinoid syndrome: episodic flushing, bronchospasm, cyanosis
- Due to serotonin (unprocessed)**
- caused by tumor spreading to liver (liver can’t process serotonin)

Polyp lining small intestine
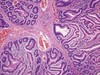
Peutz-Jeghers
Hamartomatous polyps of small bowel, (stomach, colon)
- AD inactivating mutation of LKB1 tyrosine kinase
- Pigmented skin lesions
- Branching networks of smooth muscle
- 3% develop adenocarcinoma
- Increased malignancy risk
Appendicitis- removed

Appendix- mucocele
Mucocoele: RARE
- If burst, tumor cells seed peritoneal cavity
- Malignancy with seeding
NETs: more common tumor of appendix
Large colon: polyp detected and removed

Tubular adenoma: on stalks, potentially premalignant, cancer probability depends on size
2/3 people over 65 have tubular adenomas
- 1/4 with adenomas have multiple
- Stalk: glandular epithelium
- Repetitive pattern of tubular glands
- Mucosa uninvolved
- 1% malignancy if < 2 cm
- 1/3 malignancy if > 2 cm
Pathogenesis:
- Proliferative abnormalities
- Eventual dysmaturation, impaired apoptosis
- Immature cells accumulate at tops of crypts
Cancer formation:
- Benign: high mitotic rate in all parts of glands
- Malignant: highly atypical glands; disorganized structures, cell polarity lost

Polyp removed from colon: benign or malignant? What type?

Tubular carcinoma (malignant)
2/3 people over 65 have tubular adenomas
- 1/4 with adenomas have multiple
- Stalk: glandular epithelium
- Repetitive pattern of tubular glands
- Mucosa uninvolved
- 1% malignancy if < 2 cm
- 1/3 malignancy if > 2 cm
Pathogenesis:
- Proliferative abnormalities
- Eventual dysmaturation, impaired apoptosis
- Immature cells accumulate at tops of crypts
Cancer formation:
- Benign: high mitotic rate in all parts of glands
- Malignant: highly atypical glands; disorganized structures, cell polarity lost
Removed from large colon- sessile polyp

Villous adenoma
Large broad-based cauliflower-liked lesion
- Finger-like processes with fibrovascular cores with hyperchromatic nuclei
- > 2 cm at time of discovery
Higher malignancy rates than tubular adenomas
- 50% of adenomas > 2 cm

Colon polyp

Mixed Hyperplastic polyp: variant of hyperplastic polp
Small, pale, sessile (no stalks)
- Defect in maturation: surface cells don’t slough off
- Accumulation–> sawtooth appearance
- *- Most Common (even in people < 40 years)**
- Classical type: no increased cancer risk
- *Varieties:** risk for adenocarcinoma
1. Serrated adenoma: Epithelium= serrated, hyperplastic polyp - Nuclei similar to tubular adenoma
- *2. Mixed hyperplastic**
Colon polyp

Serrated adenoma: hyperplastic polyp
Small, pale, sessile (no stalks)
- Defect in maturation: surface cells don’t slough off
- Accumulation–> sawtooth appearance
- Common (even in people < 40 years)
- Classical type: no increased cancer risk
- *Varieties: risk for adenocarcinoma
1. Serrated adenoma: Epithelium= serrated, hyperplastic polyp** - Nuclei similar to tubular adenoma
2. Mixed hyperplastic
38 year old diagnosed with colon cancer- specimen below

Familial polyposis coli:
Commonly inherited disease (50% new mutation):
- APC tumor suppressor gene mutation–> prevents degradation of beta-catenin–> poor regulation of cell proliferation–> increased E-cadherin
- Highly prone to early carcinoma development (< 40 years)
- Similar syndromes (Gardner, Turcot)= extracolonic manifestations
Thousands of tubular adenomas < 1cm
Specimen removed from 8-year-old colon

Juvenile polyps:
Hamartomas on stalks
Mutation in SMAD4 (TGFbeta pathway)
Seen in children (< 10 years), may be in adults
- Single, < 2 cm, in rectum
- Cystically dilated glands seen
- Reactive epithelial proliferation
- Increased risk for GI cancer (but not necessarily near polyp)
Removed from colon of 63 year old man with history of constipation and bleeding with bowel movements

Adenocarcinoma of colon:
Most common malignancy NOT related to smoking
- 5% of population
- Colon (L or R) and rectal cancer all have different pathogenesis, epidemiology
- Left sided= obstruct
- Right sided= anemia (insidious)
- Can see invasion through wall, metastases to local lymph nodes, liver
Most arise in adenomatous polyps
- Mutations: APC, beta-catenin, RAS (K-RAS), p53
- 15% have DNA mismatch repair defects (HNPCC)= microsatellite instability
- Defects in mismatch repair= more sensitive to chemotherapy (tumors cannot repair defect caused by drugs targeting DNA)
Appearance:
- Large, firm lesion with raised edges
- Moderately differentiate glands throughout with abundant central necrosis
- Invades muscular layers
Risk: higher in AA than whites
Risk factors: UC, Crohn disease, prior colon cancer, familial colon cancer, age
Variants seen in CRC:
Muir-Torre syndrome:
- HNPCC with skin lesions (sebaceous adenoma/carcinoma)
- Genitourinary cancers
Gardner Syndrome
- FAP with cutaneous lesions (osteomas, epidermoid cysts), desmoid tumors
Turcot syndrome:
- HNPCC + glioblastoma
- FAP + medulloblastoma
Peutz-Jeghers syndrome
- Small bowel + colonic hamartomas
- Increased GI cancer risk (CRC, small bowel, pancreatic cancer)
- Perioral pigmentation

32 year-old woman using birth control; abdominal hemorrhage

Solitary, multiple tumors
- Normal-looking hepatocytes NOT in normal architecture, 1/3 associated with hormone use (oral contraceptives)
- Benign, prone to hemorrhage

A 30 year old woman is in a car accident where she suffers abdominal trauma. At the hospital, a CT scan of her abdomen is performed and the radiologist notes a mass in her liver. A biopsy is performed to rule out a malignancy. Based on the histology below, what is her diagnosis and prognosis?

Follicular nodular hyperplasia
- *Benign** nodules with central scar, radiating fibrous bands
- non-neoplastic
- Normal appearing hepatocytes, no lobular architecture
- No association with contraceptives


HCC:
Poorly defined masses in liver
- Yellowish due to bile production
- Different stages of differentiation
- Bizarrely large cells, NO ARCHITECTURE
Symptoms:
RUQ pain
Weight loss
Early statiety
Anorexia
Jaundice
Abdominal distension
Weakness
Bleeding
Risk factors:
- Hepatitis B and C infection
- 1.5 million people in US with Hep B
- 4 million with hep C
- Annual HCC is 0.5% in asymptomatic HBV, 2.5% in symptomatic HBV
- Annual HCC in HCV= 3-8% - Cirrhosis:
- Alcoholic
- NAFLD
- Autoimmune hepatitis - Metabolic disorders:
- Herditary hemochromatosis
- Wilson’s disease
- alpha-1 antitrypsin
- Prophyria cutanea tarda - Environmental toxins:
- Arsenic
- Aflatoxin (aspergillus)
Genetics:
Necroinflammatory changes/regeneration leading to accelerated turnover–> spontaneous mutations, DNA damage
- HBV: x-protein inactivates p53 and interacts with transcriptional activation functions
- HCV: activates NFkB to stimulate proliferation, inhibit apoptosis
- *Screening**:
- Cirrhosis, alcoholics, HCV, HBV, NASH, primary biliary cirrhosis
- Serial ultrasound, AFP testing every 6-12 months
- Follow-up CT/MRI is abnormal
Paraneoplastic syndromes/Labs:
Hyperbilirubinemia
Abnormal liver function tests
Prolonged coagulation parameters
Thrombocytopenia
Hypoalbuminemia
Hypoglycemia
Erythrocytosis
Hypercalcemia
Hypercholesterolemia
Treatment:
- Sugery
- Liver transplant
- Chemoembolization

Specimen removed from liver of 2 year old child

Hepatoblastoma:
Malignant liver tumors in children 0-3 years
- V. well-circumscribed; can be large
- Mixtures of “Embryonal cells”–> ribbons/gland-like structures + fetal cells
- Trabeculae, sinusoids, primitive cartilage, mesenchymal tissues
- produce alpha-fetoprotein, sometimes hCG

57 year old man diagnosed with late stage pancreatic cancer passes away. The following is found at autopsy in his liver. What happened?

Liver metastases
Most common malignancy of liver is metastatic carcinoma
1/3 of all cancer metastasize to liver from:
- GI
- Breast
- Lung
- Pancreas
May cause massive hepatomegaly, symptoms of liver injury, impaired blood flow, jaundice
