Heme Path Flashcards
A 50 year old man comes to his physician due to increasing fatique while exercising. The physician notes that he appears pale and upon taking the history, learns that he has been chewing ice frequently. Based on his symptoms and the blood smear below, what might his diagnosis be and what could cause this? What are treatments?

- *Iron deficiency anemia**:
- Most common cause of anemia both in the US and worldwide.
- Iron deficiency leads to microcytosis due to decreased hemoglobin synthesis
- In Iron deficiency the cells are usually hypochromic in addition to being microcytic
Symptoms: ANEMIA
- Bleeding, pallor, etc
- Tachycardia, tachypnea
- Pica= craving non-food items
Causes:
Increased losses: GI bleed (most common), GU
Inadequate intake: achlorhydria (PPIs), celiac disease, IBD, autoimmune disease
- *Labs**:
- *Serum ferritin= decreased**
- Acute phase reactant: protein that increases at time of inflammation
- Storage form of iron (low= no iron storage in body)
Plus:
- Low serum iron, high total iron binding capacity, low percent saturation
Treatment:
Oral: ferrous sulfate, ferrous gluconate, ferrous fumarate
Side effects of treatment: constipation, nausea
IV: allergic reactions (less common with modern tx)
* treat underlying condition as well
Histo:
- Cigar shaped cells
- Hypochromic
- MCV usually 70s
- May be symptomatic or asymptomatic
- Platelet count may be high.
An 8 year old boy is brought to the emergency room screaming in pain. His mother says he was playing basketball and started complaining about his knees hurting. All of a sudden he was doubled over in pain and refused to move. Upon arrival in the ER labs are run and th following is seen in his blood smear. Additionally his spleen is grossly enlarged and tender. What is his diagnosis and treatment?

- *Sickle cell disease**:
- Point mutation GAG –> GTG (Position 6 glutamic acid to valine)
- Heterozygous (sickle trait) usually benign
- Homozygous causes sickling under hypoxic conditions
Symptoms:
- Spontaneous cell lysis and RBC turnover
- Increased thrombosis/infarction: Stroke, Pulmonary infarction
- Chronic inflammation
Multiple secondary complications due to infarcts
- Splenic infarcts leading to asplenic state and increased risk of bacterial infection
- Joint damage
- Non-healing skin ulcers
- Retinopathy
- Nephropathy (high comorbidity for HTN)
- Severe pain
- Opiate addiction and behavioral issues
Labs:
- Usually MCV 70s or low 80s
- Not hypochromic
- *Treatment:**
- Pain killers (opiates)
- Folic Acid
- Hydroxyurea
- Transfusion
- Bone marrow transplant
- Anti-coagulants
- Anti-inflammatories
- *Histo**: Sickle shaped red cells, pointed at both ends, caused by molecular aggregation of hemoglobin S
- Front: Howell-Jolly bodies (seen in asplenia)
- Sickle cell disease, not present in sickle cell trait
- Caused by a point mutation in b-globin chain
- Mutated b-globin polymerizes with low oxygen
- Cause changes of red cell shape

A 25 year old man comes to the doctor after a new onset of jaundice and darker urine. Initially his diagnosis was cholestasis, but he has palpable splenomegaly. Upon further interviewing, he states that something similar used to happen to his father until he passed away. Based on the blood smear below, what is his diagnosis and treatment?

- *Hereditary spherocytosis**= membrane abnormality
- Small cells without central pallor
- MCV
- May have large RDW
Other forms of membrane abnormality:
- Hereditary elliptocytosis
- Stomatocytosis
- *Histo**: Smaller, round shaped red blood cells which lack central pallor. Due to:
- Acquired immune hemolytic anemia
- Post transfusion
- Hemolytic anemia due to oxidant drugs
- Hemolysis due to a large spleen
- Hereditary spherocytosis
Hereditary Primary membrane disorder
- Spectrin Deficiency
- Mild-Moderate= Autosomal dominant
- Severe= Autosomal recessive - Beta-Spectrin - 4.1 Interaction
- Autosomal dominant
Pathophysiology:
- Cytoskeletal Abnormality
- Membrane Instability
- Membrane Loss
- decreased SA/V ratio (Spherocytosis)
- increased Osmotic fragility
- decreased RBC Deformability - Splenic Trapping
- Hemolysis
Symptoms:
- Chronic Anemia: Pallor, Jaundice, Dark Colored Urine, Splenomegaly, Cholelithisis (Gall Stones)
- Crises: Aplastic, Hyperhemolytic
Diagnosis:
- Family History
- Anemia
- Spherocytosis
- Reticulocytosis
- increased Osmotic Fragility: % hemolysis curve shifted to left: cell already spherical, less room to expand in hypotonic solution
- decreased RBC Deformability
Treatment:
Symptomatic
Splenectomy- cures the disease- red cells normalize


- *Thalassemia**:
- Usually very low MCV
- Hypochromic
- Range of symptoms from completely asymptomatic to transfusion dependent
- May have normal RBC
A 33 year old pregnant woman goes to her doctor due to increasing fatique and recurrent candidiasis. The doctor is not extremely concerned as these are normal findings during pregnancy, but upon physical exam, she notes that the woman’s tongue is very smooth and seems to be missing papilla. What condition might this woman have, what are labs that would confirm this, and what is her baby at risk for developing due to this condition?

- *Folate deficiency–**> can be due to B12 deficiency
- More likely due to poor intake of folic acid (alcohol, nutrition)
- Serum folate vs RBC folate
Symptoms:
Hematological:
- Anemia (fatique, dyspnea, SOB, syncope, chest pain)
- Leukopenia (recurrent infections)
- Thrombocytopenia (bleeding)
Epithelial:
- Generally microscopic changes with macrocytosis, increased multinucleate/dying cells
- Abnormal pap smears
- Angular chelitis, glossitis
Neural tube defects, cleft palate
Vascular disease: hyperhomocysteinemia–> arterial/venous thrombotic disease
Causes:
- Inadequate intake (NOT a concern with vegans)
- Alcohol
- Autoimmune (Crohn’s, Celiac)
- Infections (tropical Sprue, Whipple’s)
- Bowel resection
- Infiltrative disorders (scleroderma, amyloid)
Situations with increased folic acid needs:
- Pregnancy
- Chronic hemolytic anemia, inflammation
- Malignancy with high growth
Increased loss of folic acid:
- Dialysis
- CHF
- Severe liver disease
Histology:
- hypersegmented neutrophils= hallmark finding of folate/B12 deficiency
- *Labs**:
- See increase in homocysteine (can’t convert to methionine), NO increase in MMA
Treatment:
Supplements (1-5 mg/day) orally until replete
- Neurological symptoms can worsen if B12 deficiency not treated first


Myelodysplasia- ringed sideroblast
- Usually presents in middle aged or older patient
- Normal or high B12 and folate
- Usually slow onset
- Bone marrow biopsy needed to diagnose
Below is a blood smear from a 47 year-old man being treated for lymphoma. What is occuring with his RBCs?

Autoimmune hemolytic anemia (AIHA): due to lymphoma; form of macrocytic anemia
Underlying etiology of AIHA
- Immune: Transfusion reaction/alloimmune, Some drug induced, Autoimmune hemolytic anemia
- Non-immune: Drug induced, Infectious, DIC, TTP/HUS, HELLP syndrome, PNH, Sickle cell disease, thalassemia, Cell membrane or enzyme deficiencies, G6PD deficiency, Toxic exposure (i.e. snakebite), Mechanical
- Usually macrocytic due to elevated reticulocyte count
- Usually has high reticulocyte count
Causes of macrocytic anemia:
Nutritional deficiencies
- B12
- FOLATE
* Both important for DNA synthesis: issues cause increased cell size
Hemolysis (usually)
Myelodysplastic syndrome
Medication related (block DNA synthesis)
- Hydroxyurea
- AZT and other anti-virals
- Phenytoin
Toxic exposures, Alcohol
Workup:
- Basic hemolysis labs: LDH, reticulocyte count, haptoglobin, peripheral blood smear, bilirubin
- Physical exam: hypersplenism, fever, hepatomegaly
- PNH screen: flow cytometry of CD55 and CD59
- Coomb’s test and cold agglutinin test
- G6PD level
- Hemoglobin electrophoresis
- DIC panel
- Evaluation for other etiologies (DIC, sepsis, drugs, etc)
Below: TTP (thrombotic thrombocytic purpura) causing macrocytic anemia
TTP= form of autoimmune hemolytic anemia; causes small clots to form throughout body in small vessels

A 35 year old woman comes to the office with increasing fatique and a new onset of cracking, painful skin around her mouth. In her history she states she has been a vegan for the past 5 years. A blood smear reveals the following. What is her diagnosis based on the cells and her symptoms? What would her labs reveal and how should she be treated?

Megaloblastic anemia: due to B12 deficiency
- Nuclear size increased due to issues with folate synthesis (from B12 deficiency- can’t convert MMA to succinyl coA or Homocysteine to methionine–> issues with DNA synthesis)
Symptoms:
Hematological:
- Anemia (fatique, dyspnea, SOB, syncope, chest pain)
- Leukopenia (recurrent infections)
- Thrombocytopenia (bleeding)
Epithelial:
- Generally microscopic changes with macrocytosis, increased multinucleate/dying cells
- Abnormal pap smears
- Angular chelitis, glossitis
Neural tube defects, cleft palate
Vascular disease: hyperhomocysteinemia–> arterial/venous thrombotic disease
Neurological:
- Subacute combined degeneration: degenerate dorsal/lateral white matter in spinal cord–> weakness, ataxia, parasthesias, spasticity, incontinence, paraplegia
- Dementia (progressive, irreversible)
- Psychiatric disturbances
- *Causes**:
- Inadequate intake (Vegans especially at risk)
- Pernicious anemia
- Partial/total gastrectomy
- Tropical sprue
- Intestinal stagnant loop
- lack of terminal ileum
- Diphyllobothrium latum
- Congenital IF/TC abnormality
- *Labs:**
1. Low B12/folate serum levels - lower end of normal B12 can still be symptomatic
- Serum folate can be elevated artificially- look at RBC content in RBCs
2. Decreased blood counts + macrocytosis
3. Anisocytosis + macro-ovalocytes on smear
4. Hallmark= hyper-segmented neutrophils (> 5% with 5 lobes, > 1% with 6 lobes)
5. Similar values to hemolytic anemia (due to ineffective erythropoiesis): - Elevated LDH
- Low haptoglobin
- Mild unconjugated bilirubin
- *- Homocysteine (both) and MMA (B12 only) elevations**
- *Treatment**:
1) Parenteral: - 1000 ug IM daily for 1 week
- Weekly for 1 month
- Monthly after 1 month
2) orally: 1000-2000 ug /day (maintenance after repletion complete)
3) Monitor K+ during repletion in severe anemia

What changes have occured in the RBCs below? What may have caused these changes?

Ovalocytosis: seen in B12/folate deficiency
= An elongated red blood cell with blunt end; Shape varies from slightly oval or egg-shaped to long pencil-like. due to:
- Hereditary elliptocytosis
- Smaller number can be seen in iron deficiency, thalassemia, hemoglobinopathy, and other anemia.
Labs in B12/folate deficiency:
- Low B12/folate serum levels
- lower end of normal B12 can still be symptomatic
- Serum folate can be elevated artificially- look at RBC content in RBCs - Decreased blood counts + macrocytosis
- Anisocytosis + macro-ovalocytes on smear
- Hallmark= hyper-segmented neutrophils (> 5% with 5 lobes, > 1% with 6 lobes)
- Similar values to hemolytic anemia (due to ineffective erythropoiesis):
- Elevated LDH
- Low haptoglobin
- Mild unconjugated bilirubin
- Homocysteine (both) and MMA (B12 only) elevations
A 30 year old woman recently emigrated from Africa. She has been having recurrent fevers and on physical exam has a palpable spleen. Additionally she appears pale and says she has been feeling light-headed. Based on the blood smear below, what is her diagnosis?

Malaria: causes hemolysis of RBCs–> anemia (macrocytic)
A 75 year old woman comes her physician with increasing fatigue. Thyroid function tests are normal but her iron levels are on the low end of normal, and her ferritin levels are elevated. Based on the smear below, what is her diagnosis and treatment?

- *Myelofibrosis**= infiltration/failure of bone marrow due to scarring–> leads to **normocytic anemia
- Primary myelofibrosis= idiopathic**
- Secondary myelofibrosis= drug, infection, myeloproliferative disorders (below)
Also seen in:
- Hematologic malignancy= Leukemia, Multiple myeloma, Lymphoma
- Solid tumor
- Scar tissue= Myelofibrosis (primary or secondary), HIV
- Aplastic anemia (primary or secondary)
- Pure red cell aplasia (parvovirus)
Histo: strangely small cells, immature cell released prematurely by bone marrow. Can sometimes see “tear drop” cells on peripheral smear
- *Labs**:
- Iron: low to normal Fe, low TIBC, normal % saturation, elevated ferritin
- ESR/CRP may be helpful
- Elevated erythropoietin
- *Treatment**:
- Iron supplementation for low-normal levels (IV iron best)
- Epo for symptomatic chronic anemia- risk of thrombosis (should not be used with hemoglobin > 10)
Below: bone marrow infiltrated with metastatic melanoma: anemia of chronic disease due to undiagnosed malignancy

What is going on in the blood smear below of a patient with new onset anemia?

AML with phagocytosis: see cancer cells actually consuming RBCs
Below: Multiple myeloma

A 30 year old man presents to his physician after having a physical that revealed anemia and a low reticulocyte count. A few weeks ago he had a flu-like illness that has since resolved. What could be causing the changes in his bone marrow (below) and blood? In what other patients might these changes be seen?

- *Aplastic anemia**= complete absence of cells in bone marrow (visible with naked eye)
- Rare, serious condition
- Due to parvovirus, HIV, hepatitis, parasitic infection
Below: Chemotherapy anemia (meds/chemo destroy bone marrow tissue–> can also cause defective/absent RBC synthesis


Polychromasia= increase in reticulocytes

Elliptocyte
AKA ovalocyte: An elongated red blood cell with blunt end; Shape varies from slightly oval or egg-shaped to long pencil-like. due to:
- Hereditary elliptocytosis
- Smaller number can be seen in iron deficiency, thalassemia, hemoglobinopathy, and other anemia.
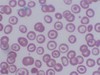
- *Target cell**: A dense central area surrounded by a relatively clear area and a peripheral rim of hemoglobin due to:
- Thalassemia
- Sickle cell disease (esp. hemoglobin C disease)
- Liver disease
- Post splenectomy
- Iron deficiency
HALT: HBC disease, Asplenia, Liver disease, Thalassemia

Echinocyte (Burr cells) – short, evenly space spicules and preserved central pallor
Due to:
- Uremia
- Bleeding ulcer
- Gastric cancer
- Artifact
- *- Distinguish from Acanthocyte (Spur cell)- mostly seen in lipoproteinemia (Liver failure)**
Due to hypophosphatemia, hypokalemia (kidney, gastric bleeding), hyperuremia (kidney)

- *Schistocyte** – Distorted, fragmented cells with 2 to 3 pointed ends
- Microangiopathic hemolytic anemia (DIC, TTP)
- Severe burns
- Prosthetic heart valves

- *Teardrop cells**= Distorted, drop-shaped cell
- seen in Myelophthisis – bone marrow fibrosis caused by various etiologies such as primary myelofibrosis, metastatic carcinoma etc.

- *Rouleaux cell**= cell aggregates resembling stack of coins, caused by increased paraprotein in serum
- Paraproteinemia = monoclonal or polyclonal gammopathy
- could also be due to Artifact – thick smear

- *RBC agglutination**= cell clumping
- Cold agglutinin disease
- Mostly IgM against I/i antigens on red cells
- Not reactive at body temperature, maximum reactivity at 4°C
- May cause extravascular or intravascular hemolysis
- Could also be due to artifact

- *Howell-Jolly bodies**= small, discrete basophilic dense inclusions usually single, nucelar remnants due to:
- Post-splenectomy
- Hemolytic anemia
- Megaloblastic anemia

- *Basophilic stippling** – Punctate basophilic inclusions, precipitated ribosome RNA due to:
- Various anemia – fine stippling
- Thalassemia – coarse stippling
- Lead intoxication – coarse stippling
TAIL: Thalassemia, Anemia, Iron deficiency, Lead poisoning

- *Microangiopathic hemolytic anemia**=
- Include thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), hemolytic uremic syndrome, uremia with hypertension, sickle cell anemia with pulmonary emboli.
- Red blood cells are fragmented by intravascular fibrin deposit in TTP and DIC
- Red cell morphology includes helmet, burr, acanthocyte, spur, spiculated, fragmented, pinched etc.

Normal segmented neutrophil
50-65% of leukocytes
Segmented nucleus (3-4 lobes)
Granular, pale pink cytoplasm
Circulate only briefly (12 hours)
Released into blood at band stage
Recruited into tissues (acute inflammation)

Neutrophilia:
Absolute neutrophil count > 7,000/ml
Etiology:
1) Infectious diseases (especially bacterial)
2) Acute stress (trauma, recent surgery)
3) Acute tissue necrosis (acute MI)
4) Medications (steroids, lithium, growth factors)
5) Pregnancy (third trimester)
6) Underlying malignancy (tumor products)
Pathophysiology:
- Increased mobilization of neutrophils from
1) Bone marrow storage pool
2) Marginal pool of circulating blood - Increased bone marrow production secondary to colony stimulating factors
Morphologic findings:
Non-specific in terms of etiology
1) Left shift (increased number of bands) (front)
2) Toxic granulation (increased primary granules)
3) Döhle bodies (blue cytoplasmic inclusions, aggregated rough ER- below)
4) Vacuolization


Neutrophilia: toxic granulation
Absolute neutrophil count > 7,000/ml
Etiology:
1) Infectious diseases (especially bacterial)
2) Acute stress (trauma, recent surgery)
3) Acute tissue necrosis (acute MI)
4) Medications (steroids, lithium, growth factors)
5) Pregnancy (third trimester)
6) Underlying malignancy (tumor products)
Pathophysiology:
- Increased mobilization of neutrophils from
1) Bone marrow storage pool
2) Marginal pool of circulating blood - Increased bone marrow production secondary to colony stimulating factors
Morphologic findings:
Non-specific in terms of etiology
1) Left shift (increased number of bands)
2) Toxic granulation (increased primary granules)- front image
3) Döhle bodies (blue cytoplasmic inclusions, aggregated rough ER)
4) Vacuolization below


Leukemoid reaction= LAP (leukocyte alkaline phosphatase) elevated
- *Differential diagnosis from CML**:
1) Stages of myeloid cells present
2) Alkaline phosphatase activity
3) Morphologic findings (toxic changes)
4) Basophilia
5) Philadelphia chromosome (BCR-ABL) - *Below**: Chronic myeloid leukemia: note cells at ALL stages of maturation, basophilia, increased platelets, and absence of toxic changes). Contrast with previous slide.
- LAP depressed


- *Neutropenia**= Absolute neutrophil count < 1800/ml
- Increased susceptibility to infection as neutrophil count drops below 1000/ml
- Agranulocytosis - virtual absence of neutrophils (depletion of blood and marrow storage pools)
- May need to use antibiotic prophylaxis
- *Pathophysiology**:
- Decreased marrow production (aplastic anemia, viral suppression, drug-related, Kostmann syndrome, cyclic neutropenia)
- Ineffective marrow production (megaloblastic anemia, myelodysplasia)
- Increased peripheral destruction (antibody mediated, overwhelming infection, hypersplenism)
Morphologic abnormalities:
Nuclear segmentation defects
1) Pelger-Huët anomaly (hyposegmented)
- inherited (autosomal dominant) - functions normally
- acquired (myelodysplasia)
2) Hypersegmentation (more than 5 lobes)
- megaloblastic anemia, hydroxyurea


- *Normal lymphocyte**: 25-40% of leukocytes (higher in children)
- Round/oval non-segmented nucleus
- Scant basophilic cytoplasm
- 80% are T cells (CD4:CD8 = 2:1)
- Large granular lymphocytes (cytotoxic T cells, NK cells)
- Function in humoral and cell-mediated immunity
- *Below**= Large granular lymphocyte= Natural killer (NK) cell or cytotoxic (CD8+) T cell
- Large granular lymphocytes can be seen in HIV, rheumatoid arthritis, clonal proliferations

A 30 year old man presents to his physician after having a physical that revealed anemia and a low reticulocyte count. A few weeks ago he had a flu-like illness that has since resolved. What could be causing the changes in his observed in the blood smear below? In what other patients might these changes be seen?

- *Lymphocytosis**= Absolute lymphocyte count > 5000/ml (>7000/ml - children, >9,000/ml - infants)
- Small, reactive mature cells–> pertussis virus
Etiology:
1) Infectious diseases (especially viral)
2) Lymphoproliferative disorders
3) Immunologic reactions (drugs, serum sickness)
Types:
- *- Small mature lymphocytes (pertussis)**
- Reactive “atypical” lymphocytes (EBV)- below (infectious mononucleosis)
1) Increased size, smudgy chromatin, may have nucleoli, abundant basophilic cytoplasm
2) Spectrum of atypical cells (CD8 T cells) - Large granular lymphocytes (HIV, rheumatoid arthritis, clonal proliferations)


Normal monocyte:
- 5-12% of leukocytes
- Irregular non-segmented nucleus
- Abundant blue-gray cytoplasm with some granules and vacuolization
- Migrate into tissues, become macrophages
- Function in acute and chronic inflammation
A 55 year old man has a 5 year history of being positive for TB- what changes have occured in his blood smear (below)?

Monocytosis= Absolute monocyte count > 800/ml
Etiology:
1) Chronic inflammatory disorders
2) Chronic infectious diseases (TB)
3) Associated with neutropenia (relative)
4) Clonal disorders (monocytic leukemias)

Normal eosinophil
3% of leukocytes
Segmented nucleus (2 lobes)
Numerous orange-red cytoplasmic granules (basic proteins)
Migrate into tissues (mucosal surfaces)
Function in allergic reactions, parasitic infections

Eosinophilia= Absolute eosinophil count > 350/ml
Etiology (specific growth factors - IL-5):
1) Infectious diseases (tissue parasites)
2) Allergic reactions
3) Asthma
4) Collagen vascular diseases
5) Neoplastic processes
- *Below**: Myocardial tissue infiltrated by eosinophils
- *Hypereosinophilic syndrome**:
- Persistent eosinophilia (> six months) with no apparent underlying cause
- Eosinophil count often > 1500/ml
- Eosinophils have abnormal morphology
- Tissue infiltration (heart, lungs, CNS)
- Treat with steroids and/or chemotherapy


- *Normal basophil:**
- Up to 1% of leukocytes
- Segmented nucleus
- Numerous purple cytoplasmic granules (inflammatory mediators, e.g. histamine)
- Distinct cell from mast cell (tissue cell)
- Immediate type hypersensitivity

- *Functional leukocyte defecit: Chronic granulomatous disease:**
1) X-linked deficiency of NADPH oxidase
2) Impaired respiratory burst and H2O2 production
3) Recurrent bacterial infections (especially catalase-positive organisms)
A 24 year old man of Mediterranean descent presents to his physician for a physical. On exam the physician notes some discrete hepatosplenomegaly. The patient reports intermittent fatique and pallor on exercise. The physician performs a blood draw which reveals a low homeglobin (7) and a smear (below). What might his diagnosis be and how should he be treated?

Beta thalassemia: Genetic abnormality leading to incomplete/absent/abnormal hemoglobin protein
Multiple mutations can cause:
- Premature stop codon
- Abnormal protein
- Protein can be absent (thal0) or altered
3 Variants of beta-thalassemia
- Beta Thalassemia minor= Single gene mutation
Symptoms: Mild, asymptomatic anemia
2. Homozygous thalassemia+
- β thal/hgb E
- Other mixed presentations
Symptoms: May have hb in 5-10 range, some skeletal abnormalities, hepatosplenomegaly
- Treated symptomatically with transfusion
- Thalassemia major (Cooley’s anemia)= Absence or severe underproduction of both β -globin genes
- Fatal early in life unless treated with transfusion
Symptoms:
- Skeletal abnormalities and hepatosplenomegaly
- Iron overload leads to death in adolescence unless treated. Accumulates in various tissues: Cardiac, Liver, Pituitary, etc.
Treatment:
- Bone marrow transplant has been used but is severely limited due to toxicity
- Hydroxyurea to increase fetal globin production has not been consistently successful
- Splenectomy can be helpful to increase lifetime of transfused RBCs
- Iron chelation therapy (improves outcomes- similar to intermedia)
* In cases of blood transfusion, chelation therapy improves outcomes (decreases iron overload)
A 20 year-old college student of Mediterranean descent goes to her physician to recieve medications to prevent malaria before leaving for a trip to Africa. After taking the prescribed medication for a week, she develops some jaundice and begins noticing blood in her urine (hematuria). She goes back to the physician who notes diffuse splenomegaly. A blood draw is performed and the following s found. What is the student’s underlying disorder and how should she be treated?

Alpha thalassemia: mutation in hemoglobin protein gene causing abnormal/decreased protein formation. 2 phenotypes:
1. Alpha thalassemic trait: two genotypes:
- Heterozygous alpha-Thal-1 (2 on one chromosome, null on other)
- Homozygous alpha-Thal-2 (one on each chromosome)
- Usually asymptomatic; decreased hemoglobin
- Rarely needs treatment
- If gene deletion on same chromosome (heterozygous), risk of hydrops in children
- *2. Hemoglobin H trait:** Absence of 3 alpha-hemoglobin genes results in very little α-globin allowing formation of β-globin tetramers
- Hemoglobin H is useless as carrier of oxygen
- Tendency to precipitate
Symptoms: moderate anemia, hemolysis, sensitivity to oxidative stress
Tx: transfusion, splenectomy, iron chelation
A 35 year old African-American male goes to see his physician before leaving on a mission to Africa. He was put on Primaquine to prophylax against malaria and has developed a sudden onset of blood in his urine as well as jaundice. His blood smear reveals the following. What is his disorder and what caused these symptoms to appear?

G6PD deficiency: See Heinz bodies within RBCs
Cytoplasmic enzymopathy (extrinsic hemolytic disorder) - Sex-linked (mostly males, some homozygous females)
* Most common enzyme defect
- *Type A= Africans (African descent)**
- Usually present when exposed to oxidant stress, medications (primaquine, dapsone, nitrofurantoin), acidosis, infection
Type B= Western
- Presents during oxidative stress, fava beans
Clinical features:
- Acute intravascular hemolysis
- Hemoglobinemia (pink-brown plasma), hemoglobinuria, jaundice (1-3 days after drug exposure)
- Severe cases= abdominal or back pain
- Heinz bodies in red cells, bite cells, blister cells, spherocytes
A 37 year old woman is pregnant with her third child. She goes to the obstetrician for a check-up and it is discovered that the fetus has died in utero due to internal hemorrhage. She has a delivery to expell the fetus (image below). What happened during the pregnancy and what tests should have been run?

Erythroblastosis fetalis= internal hermorrhage, edema, heart failure caused by maternal IgG antibodies against fetal Rh factor.
Mother should have been tested and treated after her first/second pregnancies with Rhogam to ensure she did not develop antibodies to Rh factor (she is Rh negative, the father is Rh positive–> fetus is Rh positive)
Treatment:RhIG= Rh immune globulin:
- Decreased incidence of HDN due to anti-D to < 0.1% of Rh-incompatible pregnancies
- ONLY effective in Rh-negative mother naive to D antigen (alloimmunized mothers have no benefit)
- Given in 3rd trimester, again at delivery

