Kidney Water Flashcards
Benign tumors of the kidney
List 3
Renal papillary adenoma
Angiomyolipoma
Oncocytoma
Malignant tumors of the kidney
List 3
Renal cell carcinoma
Wilms tumor
Urothelial (transitional cell) carcinoma of renal pelvis
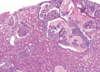
Renal Papillary Adenoma
Benign or Metastatic?
Gross Pathologic features (3):
Microscopic features (3):
Benign
Gross: Small (<1.5cm); Pale, yellow gray, Discrete, well circumscribed.
Micro: Papillary or tubular architecture; bland nuclei, no atypia, No fibrous capsule or desmopastic response

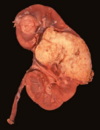
Angiomyolipoma
Benign or Metastatic?
Gross Pathologic features (3):
Microscopic features (3):
Benign
******Associated w/ tuberous sclerosis
patients may present w/ spontaneous hemorrhage
Gross: Tan to brown; Often yellow fat content; focal hemorrhage
Micro: Blood vessels; Smooth muscle; Adipose tissue

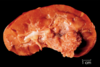
Oncocytoma
Benign or Metastatic?
Gross Pathologic features (4):
Microscopic features (3):

Benign
Gross: Well circumscribed; Homogenous; “Mahogany brown” color; Centra; stellate scar; Can be large (12cm)
Micro: Cells arranged in nests; Eosinophilic (High [mit.]); Bland/round nuclei

Renal Cell Carcinoma
Begnin or Malignant?
Pathophysio to why this is dangerous?
3 Treatments (think based on size)
Malignant
****85% primary renal malignancies
Orgin in renal cortical tubules –> metastases –> lung/bone
Txt: Partial nephrectomy; Radical nephrectomy (whole kidney); Adjunct chemotherapy (VEFG/tyrosine kinases)
Renal Cell Carcinoma survival rate depends on?
What are 3 ways to classify RCC?
Depends on stage
Avg = 5 yrs
Kidney - 95%: Distant metastases - <10%
Clear cell; Papillary; Chromophobe
What specific chromosomal abnormalities lead to Clear-cell type and Papillary type RCC?
Explain pathopsio of each
Clear-cell: Deletion Chromosome 3p (VHL gene) –> Loss of tumor suppressor gene –> promotes tumor angiogenesis thru VEGF
Papillary: Trisomy Chromosome 7 –> mutation of MET proto-oncogene (encodes tyrosine kinase receptor)
Renal Cell Carcinoma
Age it generally affects?
Gender?
Classic triad of symptoms (3):
What does RCC secrete as a tumor?
Adults > 50yo
Males > Females
- Costovertebral angle pain
- Palpable mass
- Hematuria (most common symptom)
Polycthemia: Paraneoplastic syndrome; due to secretion of erythropoietin by tumor cells.

Renal Cell Carcinoma

Clear Cell RCC
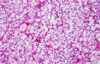
Clear Cell RCC

Papillary RCC

Papillary RCC

Chromophobe RCC
Grade of RCC


Pattern of RCC spread
- thru what gross structures of kidney and in the body?
Invasion through renal capsule into perinephric fat
Invasion into renal vein w/ proximal spread along inferior vena cava
Lymph nodes
Distant mets: lungs, bone

Wilms tumor (Nephroblastoma)
Begnin or malignant
age
Chromosome affected
what 2 syndromes is associated w/ this
Malignant
2-5yo
Mutation of WT1 gene on short arm of Chromosome 11
Associated w/ WAGR syndrome: Wilms tumor, Aniridia (absent iris), Genital anomalies, mental Retardation & Denys-Drash (Wilms tumor, gonadal dygenesis, early-onset nephropathy w/ renal failure)
How does Wilms tumor clinically present?
What does prognosis depend on?
presents as abdominal mass and abdominal pain; hematuria, intestinal obstruction, hypertension; 5-10% bilateral
.
Prognosis depends on the degree of anaplasia of the tumor cells (defined by pleomorphism, hyperchromatism, abnormal mitoses), and the stage of the tumor at time of resection. Anaplastic tumors are more aggressive.
Gross pathologic features of Wilms tumor?
Nodular
Gray to tan-white
Soft, friable, fleshy

Wilms tumor Microscopic features (3)
- Triphasic pattern*
- Primitive blastema (small/dark undifferentiated cells)
- Epithelial component (abortive tubules/glomeruli)
- Stroma (Fibrous or myxoid patterns; may contain mesenchymal elements (cartilage, muscle, bone)

Wilms tumor microscopic features (3):
Triphasic pattern
- Primitive blastema (small/dark undifferentiated cells)
- Epithelial component (abortive tubules/glomeruli)
- Stroma (Fibrous or myxoid patterns; may contain mesenchymal elements (cartilage, muscle, bone)


Wilms Tumor
What is the significance of this in WIlms Tumor?
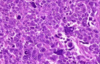
ANAPLASIA
Determines the Prognosis of Wilms tumor
- Pleomorphism, hyperchromatism, abnormal mitoses –> more aggresssive; higher resistance to chemotherapy
- Stage matters also w/ Prognosis*
Urothelial (transitional cell) Carcinoma
orgin
occurs in
associated w/
presenting symotoms
orgin in the urothelium lining the renal pelvis
adults
Associated w/ urothelial carcinoma or dyplasia elsewhere in urinary tract (“Field effect”)
Symotoms: Hematuria, urinary obstruction, hydronephrosis, flank pain
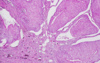
Urothelial carcinoma Gross pathological features (2):
Papillary: Exophytic mass w/ fronds
Flat: Reddened or granular appearance

Urothelial carcinoma Patho Micro
diffence in invasivness b/w papillary and flat
grade difference?
Papillary: vascular cores; lined by malignant urothelial cells; low or high grade
Flat: No paillary growth; cells disordered; Non-invasive/high grade - “carcinoma in situ”
What is the incidence of blatter cancer
Gender
Age
Causes
Clinical Signs
Males > Females
50-80
Cigarette smoking (**Most important); Chemical carcinogens (napthylamine); Infectious agent (Schistosoma haematobium *Egypt/Sudan) - assciated w/ SCC
Hematuria
What are the ways that urothelial carcinoma is diagnosed?
Hematuria, dysuria
Diagnosed with urine cytology: less invasive; cannot easily diagnose low grade malignancy
Cystoscopy with biopsy is used to more definitively diagnose, however it is more invasive
How are neoplasms of the urinary bladder are staged according to the AJCC criteria?
T: presence and extent of invasion into the bladder wall, involvement of adjacent structures
N: Presence/absence of lymph node metastases
M: Presence/absence of distant metastases
T1: invaded lamina propria
T2: Invaded muscularis
T3: Invaded soft tissue

Describe the various treatment options for bladder cancer and the indications for their use
Transurethral resection: appropriate for low grade, non-invasive papillary lesions
Bacillus Calmette-Guerin (BCG): attenuated form of TB, topically administered. Used for high grade, non-invasive lesions, carcinoma in situ. Immunotherapy: incites granulomatous inflammatory response.
Radical cystectomy: For tumors invading the muscularis (T2) or more, or carcinoma in situ not responsive to BCG
Chemotherapy: for advanced cases
Intracellular fluid (ICF): Contains ____ of TBW
2/3
Extacellular fluid (ECF): Contains ____ of TBW
1/3
The Extracellular fluid (ECF) is subdivided into 2 compartments:
Intravascular fluid (1/4 ECF)
Interstitial fluid (3/4 ECF)
What is the difference b/w plasma tonicity and osmolality?
Plasma tonicity reflects concentration of solutes that do NOT easily cross cell membranes (i.e. most sodium salts) and thus affects distribution of water between cells and ECF
Plasma osmolality includes the osmotic contribution of urea (an ineffective osmole since it moves across the cell membrane and has little effect on water movement across the cell membrane). Ethanol is another osmole that enters cells rapidly and thus has no tonicity
What is obligate osmolar excretion
- Obligate osmolar excretion: Amount of osmoles which need to be removed by the kidney in order to maintain osmolar homeostasis.
- Obligate osmolar excretion is dependent on the dietary intake a. Basal metabolism (fasting) - approximately 7 mosmol/kg/day
- Normal individuals can dilute urine to 50 mosmol/L and concentrate to 1,000 mosmol/L
- This allows a range of urine output of 7 ml/kg/day to 140 ml/kg/day a. This capacity allows us to accomplish both osmolar and water balance simultaneously
Requirements needed for excretion of a maximally dilute urine.
MAX dilute (50-75mOsmol/kg H20):
1. Delivery of solute and water to diluting sites
- fucked up in renal failure/ volume depletion
2. Proper function of the diluting segment
- Osmotic diuretics (no reabsorption in TAL)/ loop diuretics (block Na/K/2Cl)
3. AVP/ADH must be absent for the collecting duct to be impermeable to water
-needed to concentrate urine –> duhhhhhhhh bitches
Requirements for maximally concentrated urine
-
To retain significant free water (i.e. maximally concentrate the urine to 1000- 1200 mOsmol/Kg), the following are needed:
a. Development of a concentrated medullary interstitium by solute reabsorption in the TAL of the Loop of Henle
b. Presence of AVP/ADH to stimulate insertion of AQP2 into the apical membranes of collecting duct cells
d. Ability of collecting duct cells to respond to ADH/AVP by insertion of aquaporin channels
Explain the difference between osmotic and nonosmotic regulation of AVP/ADH (arginine vasopressin/anti-diuretic hormone)
Osmotic control: has a set point near normal value of serum osmolarity and is very sensitive to changes > baseline
non-osmotic: ineffective until intravascular volume > 10% baseline –> release AVP (overtakes osmotic control; activated V1A-R –> raise BP; V2-R retain H20 –> restore intravascular volume.
What are the effects of hyponatremia on the brain?

Hypertonic hyponatremia (Posm > 290 mOsmol/Kg)
Why does this occur
What leads to this
treatment
Results due to the presence of another effective osmole that causes free water to move from the intracellular compartment to the ECF resulting in cell dehydration.
- Mannitol, glycine, marked hyperglycemia
- txt: correcting the underlying condition (i.e. treatment of DKA) or removal of the osmotic agent
Isotonic hyponatremia (Posm = 275-290 mOsm/Kg)
why does this occur
Results from a laboratory artifact due to marked hyperlipidemia or hyperproteinemia
- Marked elevation in serum lipids or proteins causes a reduction in the fraction of serum that is water and results in an artificially low serum Na+ concentration
- Laboratories that use ion-specific electrodes and direct potentiometry avoid the misdiagnosis of hyponatremia
Hypotonic hyponatremia
why does this occur?
What are the 2 main classification systems
True physiologic hyponatremia that results from excess water either due to AVP/ADH stimulation or impaired water excretion
According to AVP/ADH levels
- Circulating ADH levels are appropriately elevated
- Circulating ADH levels are inappropriately elevated
- Circulating ADH levels are appropriately suppressed
According to the patients volume status
- Hypovolemic hypotonic hyponatremia
- Euvolemic hypotonic hyponatremia
- Hypervolemic hypotonic hyponatremia
Filtration
formation of a cell- and protein-free plasma filtrate in the glomerulus.
Reabsorption
movement (transport) of a substance out of the tubular lumen
Secretion
Movement (transport) of a substance into the tubular lumen
Excretion
Elmination of a substance from the body in the final urine
Describe FULL blood flow thru the kidney
All blood flows thru GLOMERULI
abdominal aorta –> renal a. –> interlobar a. –> arcuate a. –> interlobular a. –> afferent arteriole –> glomerulus –> efferent arteriole –> postglomerular capillary bed (vasa recta in medulla and peritubular capillaries in cortex) –> venules –Interlobular v. –> arcuate v. –> interlobar v. –> renal v.
important characteristics of renal vasculature
- essentially all blood flows thru glomeruli
- inflow and outflow are arterioles (high resistance)
- There are 2 capillary beds (filtration=glomerular capillaries); (absoprtion=postglomerular capillaries)

What is ultrafiltration
what is its composition like
what does it pass through grossly
The formation of a nearly protein-free filtrate of plasma as blood passes through the glomerular capillaries
- The glomerular ultrafiltrate has a composition identical to plasma except for the almost complete absence of protein.
- The ultrafiltrate is formed as fluid passes through the walls of the glomerular capillaries and into Bowman’s space to PCT
How is the filtration barrier determined?
what 3 factors compromise it
Molecular Size: Freely filtered=urea, glucose, inulin; ALBUMIN cannot pass (size and radius)
Electrical Charge: Negative charges cannot pass more readily than neutral
Molecular Shape: deformable structures can pass; globular=steric hindrance
enothelial fenestrations, basal lamina, filtration slits (space b/w pedicels)

What is more important characteristic of ultrafiltration barrier?
electrostatic restriction plays a prominent role in limiting albumin transit across the filtration barrier
Most glomerular diseases compromise both the size- and charge-selective properties of the filtration barrier
proteinuria = glomerular injury
Describe the Starling forces that influence fluid movement across capillary walls
Net Filtration Pressure = PGC–PBS–πGC+πBS
Glomerulus:
Glomerular hydrostatic pressure is high, at about 50 mm Hg, and is constant throughout the glomerulus.
Glomerular oncotic pressure increases throughout the length of the glomerulus, due to loss of plasma but retention of proteins (increased protein concentration; increased pull on water)
Bowman’s space hydrostatic pressure is about 15 mm Hg, and is constant.
Bowman’s space oncotic pressure is 0, because there is no protein content to pull water in.
The net force is 10 mm Hg in favor of filtration. This is similar to non-renal capillaries, but the filtration coefficient is much higher in the kidneys due to greater surface area and hydraulic conductivity. The forces always favor filtration in the glomerulus; never absorption.
Post-glomerulus capillaries:
Capillary hydrostatic pressure is lower than in the glomerulus, due to the pressure drop that occurs at the efferent arteriole. The pressure is about 20 mm Hg.
Capillary oncotic pressure begins at the same level as leaving the glomerulus since it relies on protein concentration. This number is high compared to systemic blood. As water is absorbed in the capillary bed, this pressure will fall.
Interstitial hydrostatic pressure is low, at about 8 mm Hg. Interstitial oncotic pressure is also low, at about 6 mm Hg.
The net movement is into the capillary, i.e. absorption, along the entire capillary bed.
Predict how changes in afferent arteriolar resistance or efferent arteriolar resistance influence renal blood flow (RBF) and glomerular filtration rate (GFR).
Increasing afferent arteriolar resistance decreases the flow through the glomerulus (RBF). This reduces the hydrostatic pressure in the glomerulus and leads to a decrease in GFR.
Decreasing afferent arteriolar resistance increases the flow through the glomerulus (RBF). This increases the hydrostatic pressure in the glomerulus and leads to an increase in GFR.
Increasing efferent arteriolar resistance decreases the flow through the glomerulus (RBF), but it increases the hydrostatic pressure in the glomerulus, thus GFR increases.
Decreasing efferent arteriolar resistance increases the flow through the glomerulus (RBF), but it decreases the hydrostatic pressure in the glomerulus, thus GFR decreases.

Describe hormonal and neural influences on RBF and GFR
The formation of angiotensin II (a result of renin release from the juxtaglomerular apparatus) causes vasoconstriction at both the afferent and efferent arterioles; contraction of mesangial cells decreases the capillary filtration coefficient, Kf, which has an impact on the rate of filtration (fluid movement = Kf * net filtration pressure). This decreases RBF and GFR.
Other substances may dilate or constrict the afferent and efferent arterioles. For example, NO dilates the vessels, while norepinephrine constricts them.
The afferent and efferent arterioles are innervated by the sympathetic nervous system.
As the sympathetic nervous system activation is increased, norepinephrine is released, which acts on both vessels to cause constriction. This decreases RBF and GFR.
During extreme activation of the sympathetic nervous system (i.e., shock), resistance in the afferent and efferent arteriole is so high that renal blood flow is severely reduced and GFR stops. The kidneys then do not receive oxygen and the cells die, leading to acute renal failure.

Define autoregulation of GFR and RBF, and name the two mechanisms involved in this phenomenon
Autoregulation is the process by which the kidney controls and alters GFR and RBF in response to changes in blood pressure and flow, in order to maintain GFR within a narrow range (80–140 ml/min).
Myogenic: ↑ mean arterial pressure → distend afferent arteriole → stretch afferent arteriolar vascular smooth muscle → smooth muscle cells contract → increase RA (in the face of increased pressure) → maintain constant RBF (and GFR)
TGF: ↑ GFR → ↑ flow thru tubule → ↑ flow past macula densa → Paracrine from macula densa to afferent arteriole → Afferent arteriole constricts → ↑ RA → LOW PH in glomerulus → LOW GFR

List the percentages of Na+ reabsorbed by each nephron segment
PCT
Loop of Henle (TAL)
DCT
CD
PCT = 67%
Loop of Henle = 25%
DCT = 5%
CD = <3%
How does reabsorption occur in the PCT
apical Na+ co-transporters (SGLT1/2, NaPi2) and Na+/H+ exchanger
In the proximal tubule, Na+ moves across the apical membrane of the tubule down its concentration gradient, mostly via the Na+- glucose symporter. There is also the Na+/H+ antiporter, and a Na+-phosphate symporter (important during bone development). On the basolateral membrane, the Na+,K+-ATPase pumps Na+ into the interstitial space so it can be reabsorbed by the postglomerular capillaries. Water follows the movement of Na+, as does Cl-.

How does Na+ reabsorption occur in Loop of Henle (TAL)
name 2 diuretics that work here
What syndrome occurs w/ loss of f(x) NKCC2 mutations
tDL, only water can move out of the tubule. The tDL is not permeable to Na+.
In the TAL of the loop of Henle, the Na+,K+,2Cl- transporter (NKCC2) brings sodium, potassium, and chloride through the apical membrane; this is where most Na+ enters. The Na+,K+-ATPase in the basolateral membrane then pumps out Na+ into the interstitial space. The Na+/H+ exchanger also brings in some Na+ here.
Called the diluting segment; uses a lot of energy to pump out Na+ on the basolateral side.
Diuretics like furosemide and bumetanide block NKCC2, preventing sodium and water reabsorption. They also cause increased distal sodium reabsorption (with potassium loss), so are potassium wasting. They block the TGF mechanism, preventing a decrease in GFR.
Loss of function mutations of NKCC2 causes Bartter’s syndrome, with salt wasting, hypokalemia, alkalosis, and hypercalciuria.
How does reabsorption occur in DCT
What type of diuretics work here
Loss of f(x) mutation of NCC channel leads to what syndrome
Na+ is brought in by the Na+/Cl cotransporter through the apical membrane. Na+,K+-ATPase pumps Na+ into the interstitial space through the basolateral membrane. Cl- follows passively through the basolateral membrane.
The NCC channel is sensitive to thiazide diuretics. Prevents reabsorption of Na+ and water. These diuretics are potassium wasting as well, because the collecting duct will see more Na+ and water, and will compensate with more Na+ channels (ENaC). As a result, the Na+,K+-ATPase will have to increase its activity, bringing more K+ into the cell in the process, which has the opportunity to diffuse out into the duct via a K+ channel (thus wasting).
Loss of function mutation causes Gitelman’s syndrome, with salt wasting, hypokalemia, alkalosis, and hypocalciuria.
How is sodium reabsorbed in Collecting Duct
What drug blocks the ENaC channel
What hormone is the CD sensitive too
What syndromes (2) occurs in ENaC mutation –> explain pathopsio
ENaC (epithelial Na+ channel) brings sodium through the apical membrane. The Na+,K+-ATPase pumps Na+ through the basolateral membrane to the interstitial space where it can be reabsorbed by the postglomerular capillaries.
Amiloride blocks the ENaC channel, causing more Na+ and water excretion. This diuretic is potassium sparing, since the collecting duct will not have the opportunity to detect and upregulate the ENaC channel in a way that will affect potassium excretion significantly.
The ENaC is located in principal cells and are sensitive to aldosterone; in response to binding of aldosterone to the aldosterone receptor, the ENaC channel will be produced and shuttled to the apical surface and to allow reabsorption of Na+.
In Liddle’s syndrome, a mutation causes the channel to always be open. This leads to salt retention and severe hypertension.
In Pseudohypoaldosteronism, a mutation causes loss of function of the channel. This leads to salt wasting, hyperkalemia, acidosis, and hypotension.
Describe the tubular reabsorption of glucose. Explain how diabetes mellitus or defects in glucose transport can result in excretion of glucose
In early PCT, glucose is reabsorbed by SGLT2 (Na+, glucose cotransporter; high capacity, low affinity, *98% glucose reabsorbed) –> Basolateral GLUT2 –> diffusion to intersitial space
In the late PCT, the apical SGLT1 (2Na+, glucose cotransporter; low capacity, high affinity for glucose –> Basolateral GLUT1 channels facilitate diffusion to the interstitial space.

What compounds are secreted by the PCT
Organic -: Phenol red; PAH (diagnostic agent measuring RPF); Penicillin; Furosemide; Acetazollamide; Creatinine
Organic +: Histamine; Cimetidine; Cisplatin; NE; Quinine; Tetraethylammonium; Creatinine

Podocytes form the ______ layer of the Bowman’s capsule
visceral
The glomerular basement membrane lies b/w what 2 structures?
What makes the GBM?*
fenestrated endothelium of the glomerulus and the podocyte processes that wrap around the capillaries.
secreted by podocytes*
What type of epithleium lines the calyces, renal prelvis, ureters, urinary bladder, proximal urethra
Transitional epithelium
renal pelvis
acts as a funnel that combines all the major calyces and narrows the tube to connect to the ureter
calyx
part of the urinary tract that collects urine from the medullary pyramid and delivers it to the ureter
Ureteropelvic junction
where the renal pelvis joins to the ureter; there is a narrowing at this junction. (Constriction point)
ureterovesicle junction
where the ureter connects the bladder; there is narrowing at this junction. (constriction point)
What can alter Kf
- change in surface area
- Humoral agents (Angiotensin II) decrease SA for filatration via ctrx of mesangial cells
What ions are sensed by the macula densa cells?
What happens when this is elevated
What is this called
NaCl (NKCC2)
increased [NaCl] (NKCC2) –> release of signaling molecules (Ca, ATP, Adenosine) from macula densa –> CTRX afferent arteriole –> LOW GFR
Tubuloglomerular Feedback
Which segment of the kidney has the highest osmotic H20 permeability with and without AVP present?
PCT (AQP1)
Countercurrent multiplication
responsible for?
Requirments (3)
What diuretics act to inhibit
establishing medullary interstitial osmotic gradient
Countercurrent flow
Different water permeability of adjacent structures (descending/ascending limb)
Energy (Na,K-ATPase)
“loop diuretics” - Furosemide; Bumetanide
The ____ is isosmotic and delivers roughly what mOsm/L to the loop of henle
PCT
300mOsm/L
Countercurrent exchange
What is it and where is [solute] highest?
active or passive process?
ADH effect on countercurrent exchange?
countercurrent blood flow (vasa recta)
passive
Solute diffuses from the ascending vasa recta into the descending vasa recta. This process “traps” solute in the inner medulla.
ADH decreases medullary blood flow –> Enhancing concentration gradient
How does increase/decreased blood flow through vasa recta can alter urine flow and osmolarity
Increase=the urine cannot be as concentrated and the flow increases
Decrease=increasing interstitial osmolarity and increasing ability of the countercurrent multiplier to remove water from the urine
What are the stimuli for ADH release?
Where does this occur?
Where does it act?
increase in plasma osmolality detected in supraoptic and paraventricular neurons of the hypothalamus
Right Atrium baroreceptors for large decreases >10%
Collecting Duct and Vasoconstriction of Vasa Recta
Explain the roles of ADH in regulating water balance

How does the renin-angiotensin system maintain Na+ balance?
Full Mechanism (5)

Describe the regulation of renin secretion
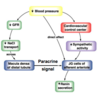
What are the actions of Angiotensin II on retention of Na+ and rest of body
think direct and indirect
Constrict renal arteriole –> Decrease GFR (decreases filtered load of Na+ –> Increases PCT Na+ reabsorption (Na+/H+ exchanger)
ANGII –> Aldosterone –> High Na+ reabsorption in CD
ANGII vasoconstriction –> Increased TPR
Stimulates thirst reflex; ADH release; Decreased medullary blood flow
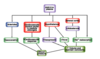
Effect of Aldosterone on Na+ reabsorption
Increased ANGII –> Aldosterone –> Increased Na+ reabsorption by CD (ENaC *Amilioride)
Triggered by increased AngII, Decreased plasma Na+, Increased plasma K+
Effect of Catecholamines on Na+
Where does it ilicit a response?
Activated by SNS
Increased Na+ reabsorption by PCT (Na+/H+ exchanger)
ANP effect on Na+
mechanism
Atrial stretch (increased ECF)
Decrease Na+ reabsorption in CD (Inhibits ENaC)
Endogenous Digitalis-like Substance
Mechanism
Increased ECF volume
Decrease Na+ reabsorption by all nephron segments
Direct effect to inhibit the basolateral Na,K-ATPase
Responses to a Spontaneous increase in GFR
Glomerulo-tubular Balance
PCT reabsorbs a constant fraction of filtered Na+ (67% of filtered load) Mechanism: ↑ GFR –> ↑ oncotic pressure of plasma entering the peritubular capillaries –> ↑ reabsorption (Starling forces).
Tubuloglomerular Feedback
↑ GFR –> ↑ solute and water delivery to the macula densa –> TGF-mediated afferent arteriolar constriction –> ↓ GFR back toward normal.
Responses to an Abrupt Increase in Na+ intake
Factors that favor an increase in GFR
↓ plasma oncotic pressure –> Starling forces –> ↑ GFR ( ↑ ECF volume dilutes plasma proteins)
↑ arterial pressure –> ↑ capillary hydrostatic pressure in the glomeruli (autoregulation is not perfect) –> ↑ GFR
↓ AngII levels –> ↓ renal arteriolar resistance –> ↑ RBF & GFR ( ↑ ECF volume –> ↓ renin release)
↓ sympathetic tone & circulating catecholamines –> ↓ renal arteriolar resistance –> ↑ RBF and GFR ( ↑ arterial pressure sensed by carotid & aortic baroreceptors)
Responses to an Abrupt Increase in Na+ Intake
Factors that decrease tubular Na+ reabsorption (to allow ↑ Na+ excretion)
↓ AngII levels –> ↓ Na+ reabsorption in proximal tubule
↓ aldosterone levels –> ↓ Na+ reabsorption in collecting duct ( ↓ AngII levels –> ↓ aldosterone release)
↓ sympathetic tone & circulating catecholamines –> ↓ Na+ reabsorption in proximal tubule
↑ ANP levels –> ↓ Na+ reabsorption in collecting duct ( ↑ blood volume sensed by atrial stretch receptors)
↑ endogenous digitalis-like substance (unknown stimulus) –> generalized ↓ in Na+ reabsorption
Distribution of K+ in the body
140 mEq/L inside the cell
A bump in potassium intake will quickly be shuttled into the cells; over time the extra potassium will be excreted in urine.
K+ handing in PCT
Loop of Henle
PCT=80% (follows H2O paracellularly)
Loop of Henle=10% (TAL NKCC2)
K+ handling in CD
difference b/w 2 areas?
α-Intercalated cells (ICT, CCT & MCD): active reabsorption (during low dietary K+ intake)
*Apical uptake via H+-K+-ATPase –> ↑intracellular [K+] –> exits cell via basolateral K+ channel
Principal cells (ICT and CCT): active secretion
*K+ uptake from peritubular interstitium via basolateral Na+-K+-ATPase –> ↑ intracellular [K+] –> passive flux of K+ across apical membrane (probably via a K+ channel)
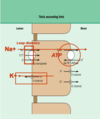
What factors determine the net rate of K+ excretion in the CD
2 mechanisms
↑ Dietary K+ intake –> ↑ [K+]ECF –> increase Na+-K+-ATPase activity –> ↑ K+ uptake across basolateral membrane of distal tubule & collecting duct cells –> ↑ K+ secretion –> ↑ K+ excretion.
↑ PK –> direct stimulus of aldosterone release (adrenal cortex) –> ↑ apical Na+ entry into CD cells –> ↑ lumen negativity and Na+- K+-ATPase activity –> ↑ K+ secretion –> ↑ K+ excretion.

K+ wasting diuretics
Furosemide
Mannitol
Where do they work??
K+ sparing diuretics
Amiloride
Spironolactone
Where do they act?
Intracellular fluid (ICF) contains ______ of TBW
2/3
Extracellular fluid (ECF) contains _____ of TBW
1/3
The ECF is divided into 2 compartments
% ECF ?
Intravascular = 1/4 of the ECF
Interstitial = 3/4 of the ECF
When would Glucose and Urea contirbute GREATLY to Posm?
When glucose is markedly elevated (uncontrolled DM) or in reduced renal function (elevated urea)
What is the fefinition of obligate osmolar excretion?
Amount of osmoles which need to be removed by the kidney in order to maintain osmolar homeostasis
*Obligate osmolar excretion is dependent on the dietary intake Basal metabolism (fasting) - approximately 7 mosmol/kg/day
Requirements for excretion of a maximally dilute urine (50-75 mOsmol/kg H2O)
-
Delivery of solute and water to diluting sites
- renal failure (low GFR) –> low solute delivery
- volume depletion or effective intravascular volume depletion (CHF, cirrhosis, nephrotic syndrome), PCT has higher Na+/H2O delivery –> low solute delivery -
Proper function of the diluting segment
- osmotic diuretics (mannitol)
- loop diuretics - AVP/ADH must be absent for the collecting duct to be impermeable to water
Requirements for Excretion of Maximally Concentrated Urine (1000- 1200 mOsmol/Kg)
To retain significant free water
- Development of a concentrated medullary interstitium by solute reabsorption in the TAL of loop of henle*
- Presence of AVP/ADH –> AQP2 in collecting duct*
- Abilitiy of collecting duct to respond to AVP/ADH by insertion of AQP2*
Nonosmotic mechanism of AVP secretion
vs.
Osmotic mechanism of AVP secretion
intravascular volume depletion (>10%) *life-saving thru V1/V2R –> raise BP/retain H20/restore intravascular volume
vs.
sensitive to any changes above baseline serum osmolarity
Hyponatremia is < _______ mEq/L
135 mEq/L
Hyponatremia leads to a great increased mortaliity (60%) in hostpitalized patients
(is a marker not a cause)
usually due to nonosmotic release of AVP
Effects of hyponatremia on the brain
- The brain is most susceptible to the sudden decrease in serum Na+ because it is confined within the rigid skull
- Acute hyponatremia causes nausea, vomiting, and confusion due to brain edema
- Severe brain edema leads to seizures, even herniation and death
- When hyponatremia develops slowly (over several days), the brain cells can adapt by releasing intracellular K+ and Cl- initially; and subsequently, organic osmolytes (myoinositol, amino acids) such that the cell volume is reduced to near normal levels
- This is the reason why chronic hyponatremia is frequently asymptomatic unless the serum Na+ is very low (i.e. <120 mEq/L)
Posm > 290 mOsmol/Kg = _______ hyponatremia
what psio causes this
Hypertonic hyponatremia
from presence of another effective osmole that causes free water to move from the ICF –> ECF –> cell dehydration
*Mannitol, glycine, High [glucose]
rx: correct the underlying condition
Posm = 275-290 mOsmol/Kg = _______ hyponatremia
what psio causes this
Isotonic or “Pseudohyponatremia”
results from a laboratory artifact due to marker hyperlipidemia or hyperprotenemia –> reduced fraction of serum that is water –> artifically low serum [Na+]
*labs that use ion-specific electrodes avoid this misdiagnosis
Posm < 275 mOsmol/Kg = _______ hyponatremia
what psio causes this
2 main classification systems
Hypotonic hyponatremia
results from excess water from either AVP/ADH or imparied water excretion
- AVP/ADH
- Volume status
Classification of hypotonic hyponatremia by volume status
psio behind this
how do patients appear
what would urine output look like (2)
True volume depletion (low ECF volume) –> loss of fluid volume (Na+) –> stimulate ADH secretion –> attempt to restore ECF
*volume depleted (hypotension, flat neck veins, orthostatic)
- GI losses; blood losses; excessive sweating, burns* –> Urine Na+ < 20 mEqL (max reabsorption at PCT)
- Renal Na+ losses* (diuretics, adrenal insufficiency, salt-wasting nephropathies) –> Urine Na+ > 20 mEqL
Euvolemic hypotonic hyponatremia
psio behind this
how do patients appear
Primary water gain (normal ECF) –> excess ADH (inappropriate), excessive water intake (psychogenic polydipsia), reduced solute intake (beer-drinkers), hypothyroidism
appear euvolemic on exam
Hypervolemic hypotonic hyponatremia
2 types
ECF volume excess w/ intravascular volume depletion (low effective circulating volume)
- heart failure, cirrhosis, nephrotic syndrome
- ADH is inappropriately activated due to low circulating volume
- Urine Osm is high due to ADH activity; Urine Na+ <20 mEq/L –> max reabsorption at PCT/renin from intravascular volume depletion
*Advanced renal failure –> high Urea –> high measured plasma osmolality –> dilutional hyponatremia
management of hyponatremia
- In chronic hyponatremia, the Na+ correction should never exceed >10 mEq/L in 24 hours to avoid osmotic demyelination syndrome
- If symptomatic with life-threatening seizures, then raising the serum Na+ by ~ 4-6 mEq/L acutely with 3% hypertonic saline is appropriate
- Correction of the intravascular volume with 0.9% normal saline is appropriate for hypovolemic hyponatremia (caution to avoid correcting too quickly; once volume is repleted, the stimulus for ADH will be suppressed and patients can have a brisk water diuresis increasing the serum Na+ quickly)
- Correction of the underlying cause (treatment of hypothyroidism, increasing solute intake for tea and toasters)
- SIADH – correct the underlying cause if identifiable otherwise free water restriction (generally to 1200 ml per day), increase solute intake (if unable to eat enough protein then salt tablets may be added), and can add loop diuretics in efforts to limit the addition of NaCl to the medullary interstitium which is needed in order to maximally concentrate the urine in the presence of ADH • V2 receptor antagonists (Conivaptan and Tolvaptan are now available but are costly and are not indicated for long-term use
- Hypervolemic hyponatremia (edematous states) are typically treated with diuretics and fluid restriction
Osmotic Demyelination Syndrome
what causes this

area in brain that are slowest in reaccumulating osmolytes after rapid correction –> massive axonal demyelination in pontine white matter secondary to osmotic changes (chronic hypnatremia)
acute parallysis, dysarthria, dysphagia, diplopia, loss of consciousness. Can cause “locked-in syndrome.”
correcting serum Na+ too fast“from low to high your pons will die”
“from high to low your brain will blow” (cerebral edema/herniation)
What risk factors predispose you do getting osmotic demyelination syndrome
- duration of hyponatremia (>2 days)
- correction of Na+ >10 mEq/L within 24h
- low serum Na+ (<120; <105 mEq/L increasing risk)
Describe the patient population who is at particular risk for hypernatremia even in the absence of specific problems with urinary concentration (2)
hypernatremia primarily occurs in patients who cannot express thirst normally (i.e. elderly, infants) OR who do not have access to free water (hospitalized patients in the intensive care unit)
Explain the difference between dehydration and hypovolemia
Loss of free water only is referred to as dehydration
Loss of both Na+ and water is referred to as hypovolemia
Describe the difference between central and nephrogenic diabetes insipidus
Central DI – due to insufficient release of ADH in response to an increased serum Na+ or osmolarity (can be partial or complete impairment of ADH).
-Due to lesions of the hypothalamic osmoreceptors, supraoptic/paraventricular nuclei, or superior portion of the supraoptichypophyseal tract due to trauma, surgery, or tumors
Nephrogenic DI (can be partial or complete)– reduced action of ADH at the collecting tubule due to either mutations in the V2R or AQP2 or medications (i.e. lithium).
Explain the risk of too rapid of correction of hypernatremia
In chronic hypernatremia (>48h duration), the serum Na+ correction should not exceed >10 mEq/L over 24h in efforts to avoid cerebral edema (rapid lowering once cerebral adaptation has occurred causes additional osmotic water movement into brain cells resulting in cerebral edema, encephalopathy, seizure, and permanent neurologic damage)
In both hyponatremia and hypernatremia, the serum Na+ must be followed closely (i.e. checked every 2-4 hours) and therapy should be modified accordingly to AVOID too rapid of correction
4 mechanisms of K+ balance
- K+ intake through the diet
- GI losses: GI tract secretes 5-10% of absorbed K+ daily
- Renal losses: 90-95% is regulated by the kidney
- Transcellular K+ shift: Overall K+ stores remain the same but can redistribute between the ICF and ECF based on transcellular shift
how is potassium is reabsorbed in the thick ascending loop of Henle?
10-25% is reabsorbed in the TAL of Henle
driven by NKCC2
active process driven by basolateral Na,K-ATPase
K+ is recycled across luminal membrane, allowing continued activation of NKCC2
Activity of luminal K+ channel is inhibited by ATP allowing a ling to level of Na+ reabsorption
- As more Na+ enters cell, Na+ will be transported out of the cell into the peritubular capillary by Na+-K+ ATPase –> lowering cellular ATP level and stimulates activity of luminal K+ channel
- Permits return of reabsorbed K into lumen and further Na+ absorption
PSIO behind how the Collecting tubule principal cell does K+ secretion
K+ actively transported into cell by Na+-K+ ATPase at basolateral membrane • Secreted into tubular fluid (TF) down a favorable electrochemical gradient via luminal K+ channels (ROMK)
Governed by factors that affect passive transport
Concentration gradient across luminal membrane
– High intracellular [K+] and low TF [K+]
Electrical gradient generated by reabsorption of Na+ via luminal Na+ channels (ENaC)
K+ permeability of luminal membrane – # of open K+ channels

4 main factors that affect K+ secretion into the tubular fluid
Aldosterone – augments K+ secretion in principal cells
– Increase # open Na+ and K+ channels in luminal membrane
– Enhances activity of Na+-K+ ATPase pump
Plasma [K+]
– Increase # open Na+ and K+ channels in luminal membrane
– Enhances activity of Na+-K+ ATPase pump
Distal flow rate – Increase in distal flow rate washes secreted K+ away and replaces with relatively K+ free fluid –> favorable [K+] gradient for secretion into TF – When distal flow rate reduced, high luminal [K+] (due to less washout of secreted K+) and low urine flow –> reduction in absolute rate of K+ secretion (additionally voltage gated channels – Maxi K – stimulated by flow)
Distal Na+ delivery – Entry of Na+ via Na+ channel (ENaC) makes lumen electronegative – Transport of Na+ into peritubular capillary by ATPase pumps more K+ into cell
– More K+ secreted into electronegative lumen
Transcellular shift Hypokameia (paslma K+ <3.5 mEq/L
causes (4)
Insulin and Beta-2 agonist increase Na,K-ATPase
Alkalosis: H+ will leave cell in order to minimize extracellular pH (to maintain electoneutrality, K+ will enter the cell; small effect)
Hypokalemic period paralysis: Characterized by acute attacks in which sudden movement of K+ into cells lowers the plasma K+ to 1.5-2.5 mEq/L
Precipitated by rest after exercise, stress, or a carbohydrate meal (events associated with release of epinephrine or insulin) a. Familial – autosomal dominant associated with mutations in dihydropyridine calcium channel in skeletal muscle b. Acquired – thyrotoxicosis (predominantly young Asian males)
GI losses Hypokameia (plasma K+ <3.5 mEq/L)
causes
Vomiting and nasogastric tube output
- Associated with metabolic alkalosis due to HCl acid loss
- K+ loss from emesis ~ 5-10 mEq/L
- Concurrent urinary losses due to activation of aldosterone and an increase in plasma bicarbonate that increases the filtered bicarbonate above its reabsorptive threshold. Because Na+ pairs with bicarbonate in the tubular fluid, the increase in distal delivery of Na+ will further promote K+ loss – net effect is profound hypokalemia
Diarrhea and laxatives
- Associated with metabolic acidosis due to bicarbonate losses
- K+ loss from stool can range from 20-50 mEq/L
Renal losses Hypokameia (plasma K+ <3.5 mEq/L)
3 main
I. Conditions associated with metabolic alkalosis
- Diuretics (loops and thiazides)
- Salt wasting nephropathies (associated with hypotension) a. Bartter’s syndrome: (*think loop diuretic) –> Defect in NaCl reabsorption in TAL of Loop of Henle (any transporter)
* Gitelman’s syndrome* (*think thiazide diuretic) Defect in the gene encoding the thiazide-sensitive NaCl cotransporter in the distal convoluted tubule *Low urinary calcium distinguishes Gitelman’s from Bartter’s - Mineralocortecoid excess (associated with hypertension)
* Liddle’s syndrome*: gain of function mutation in the epithelial Na+ channel (ENaC) located on the luminal side of the principal cell
II. Conditions associated w/ metabolic acidosis
-renal tubular acidosis/nonreabsorbable anion
III. Magnesium
Hypokalemia occurs in 40-60% of cases of hypomagnesemia 2. Often due to underlying disorders that waste both magnesium and K+ (i.e. diarrhea, diuretics)
clinical manifestations of hypolalemia
4 main categories
Cardiac arrhythmias and ECG abnormalities
- Premature atrial and ventricular beats, sinus bradycardia, AV block, and ventricular tachycardia/fibrillation 2. Decrease in amplitude of T wave and increase in amplitude of U wave (occur at end of T wave – seen in lateral precordial leads V3-V6)
Muscular
weakness and muscle cramps (hyperpolarize skm cells impair ctrx)
impair NO release –> predispose to rhabdomyolysis during vigorous exercise
Hormonal
impairs insulin release and end-organ sensitivity to insulin
Renal
tubulointerstitial and cystic changes in the parenchyma of the kindey (prolonged hypokalemia)
polyuria (impairs concentrating ability)
hypertension (increased renal vascular resistance)

Understand how the urinary potassium to creatinine ratio can help identify urinary losses from shift and GI losses
Urinary K+ to creatinine ratio: < 15 mEq/g (assuming most people excrete 1000mg of creatinine in urine) suggests appropriate conservation of K+ and extrarenal cause (GI, transcellular shift)
Metabolic acidosis
what causes low urinary K+ to creatinine ratio (<15mEq/g)
what causes high urinary K+ to creatinine ratio (>15mEq/g)
low= stool losses
high= RTA, nonreabsorbable anion
Metabolic alkalosis
what causes low urinary K+ to creatinine ratio (<15mEq/g)
what causes high urinary K+ to creatinine ratio (>15mEq/g)
low= vomiting, remote use of diuretics
high= check BP and volume status
low bp/volume= Diuretics, salt wasting nephropathies, ongoing vomiting with sustained metabolic alkalosis
high bp/volume= mineralocorticoid excess
*always check magnesium - hypokalemia due to magnesium wasting cannot be corrected until magnesium is corrected
causes of pseudohyperkalemia
Elevation in measured serum K+ is due to K+ movement out of the cells during or after a blood specimen has been drawn
- Hemolysis (destruction of red blood cells) due to technique (clenching, prolonged tourniquet, venipuncture trauma) during blood draw
- Thrombocytosis
- Leukocytosis (acute leukemia)
causes of transcellular shift hyperkalemia
Metabolic acidosis: H+ will enter the cell in order to buffer the extracellular pH (primarily inorganic acids) 1. In order to maintain electroneutrality, K+ will leave the cell (overall a small effect)
Hyperglycemia and hyperosmolality 1. Elevation in serum osmolality results in water movement from the ICF to the ECF
a. Results in increased [K+] in the cell – K+ will move out of the cell down concentration gradient
b. Solvent drag from water movement
Nonselective β-antagonists: Interfere with K+ uptake into the cell by β- adrenergic receptors
Exercise: K+ released by muscle cells (causes local vasodilatation for increased blood flow)
Tissue breakdown: Rhabdomyolysis (muscle breakdown), lysis of large tumor burden after chemotherapy, burns cause release of K+ into the ECF
Digitalis (Digoxin) toxicity: Inhibits the Na+/K+ ATPase pump vii.
Hyperkalemic familial periodic paralysis:
- Autosomal dominant due to point mutation skeletal muscle Na+ channel
- Precipitated by cold, rest after fasting, and small amounts of K+ ingestion

Renal: Decreased urinary excretion Hyperkalemia

main causes of renal failure in hyperkalemia
- Able to maintain near normal levels of K+ as long as distal flow rate and aldosterone secretion is maintained
- Hyperkalemia develops in patients who are oliguric (decreased distal flow rate) who have an additional problem a. Excess K+ load, aldosterone blockade (ACE inhibitors, angiotensin receptor blockers, and alodosterone blockers)
main causes of Volume depletion w/ decreased distal delivery of Na+ in hyperkalemia
- Hypovolemia
- Effective arterial volume depletion with extracellular volume excess
a. Heart failure
b. Cirrhosis of the liver
main causes of Functional hypoaldosteronism (either low aldosterone state or resistance to the effects of aldosterone) in hyperkalemia
(3)
-
Mineralocorticoid deficiency
- Primary adrenal insufficiency (adrenal gland does not generate aldosterone in response to renin)
- Hyporeninemic hypoaldosteronism (diabetic – Type IV RTA) – low plasma renin levels and aldosterone levels) - Tubulointerstitial disease a. Sickle cell disease and urinary tract obstruction i. Impaired Na+ reabsorption in the principal cell reducing K+ and H+ secretion (also called Type I distal hyperkalemic RTA)
-
Drugs
- ACEi, ARB, spironolactone –> decrease renin release
- Nonsteroidal anti-inflammatory drugs (NSAIDs) and beta blockers
Identify the clinical manifestations of hyperkalemia: (serum K+ > 7 mEq/L with chronic hyperkalemia or possibly lower values if rise is more acute)
a. Severe muscle weakness or paralysis
i. Ascending weakness that begins with the legs and progresses to trunk and arms –> flaccid paralysis
b. Cardiac arrhythmias and ECG abnormalities
i. Bundle branch block, advanced AV block, sinus bradycardia, sinus arrest, slow idioventricular rhythm, ventricular tachycardia and fibrillation, asystole
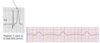
ii. ECG
1. Early - tall peaked T waves and shortening of the QT interval
2. More advanced - prolongation of PR and QRS interval (may lose P wave altogether with widened QRS sine wave)
How does Transtubular K+ gradient (TTKG) work in hyperkalemia diagnosis?
may help distinguish functional hypoaldosteronism from other disorders (volume depletion)
i. Gradient between the tubular fluid and plasma K+ - estimates aldosterone activity by measuring the K+ concentration in the tubular fluid at the end of the cortical collecting tubule (site responsible for K+ secretion)
ii. [Urine K ÷ (Urine osmolality / Plasma osmolality)] ÷ Plasma K
iii. Value < 5 is suggestive of hypoaldosteronism
Treatment of Hyperkalemia (3 approaches)
a. Antagonizing membrane effects of K+ with calcium (reserved for patient’s with ECG changes, acute rises in serum K+)
i. Calcium Choloride
b. Driving extracellular K+ into cells
i. Insulin administered with glucose
ii. β-2 agonist (albuterol)
c. K+ removal
Diuretics
Cation exchange resins
Dialysis
_*treatment of reversible causes_

Metanephric blastema
Bowman’s capsule
Tubular system: PCT, Loop of Henle, DCT
Urteric bud forms
Collecting tubules
Major/Minor Calyces
Renal Pelvis
Ureters
WT1 and PAX-2 TF mutations lead to what diseases?
WT1 transcription factor
Null mutation results in renal agenesis. Mutation results in Wilm’s Tumor (kidney cancer) = Wilm’s tumor suppressor gene 1= WT1.
WAGR associated w/ deletion of WT1
Denys-Drash associated w/ mutations of WT1
PAX-2 transcription factor
Mutations result in malformations like coloboma (and small kidneys)
POTTER sequence (syndrome)

P – Pulmonary hypoplasia (urine/amniotic fluid is needed for lung development)
O – Oligohydramnios (insufficient amniotic fluid = trigger)
T – Twisted face (compressed features due to oligohydramnios)
T – Twisted skin (ditto)
E – Extremity defects (ditto)
R – Renal failure in utero (primary cause)
Unilateral multicystic dysplasia = MCDK

Ureteric bud fails to induce differentiation of metanephric mesenchyme –> nonfunctional kidney consisting of cysts and connective tissue. Predominantly nonhereditary and usually unilateral; bilateral leads to Potter sequence.
Horseshoe kidney

Inferior poles of both kidneys fuse abnormally. As they ascend from pelvis during fetal development, horseshoe kidneys get trapped under inferior mesenteric artery and remain low in the abdomen. Kidneys function normally. Associated with hydronephrosis (eg, ureteropelvic junction obstruction), renal stones, infection, chromosomal aneuploidy syndromes (eg, Turner syndrome; trisomies 13, 18, 21)
MOA of Carbonic Anhydrase Inhibitors
Acts primarily on the PCT cells to inhibit bicarbonate absorption. Thus prevents disposal of carbonic acid, slowing the reaction that uses H+ ions, and slowing the Na+/H+ antiporter, thus preventing reabsorption of sodium. They are poor diuretics, however, because the rest of the tubule adapts to reabsorb more sodium.
chemically related to sulfonamide antimicrobial agents

MOA ‘loop diuretics’ - furosemide
inhibit the NKCC2 in the TAL of the loop of Henle by binding to the Cl- binding site –> High excretion of Na+ and Cl- w/ an associated increase in Ca2+ and Mg2+ (loss of transepithelial electropoetential difference - normally drives reabsorption
adverse effects: Higher Na+ delivery to distal tubules enhances K+ and H+ excretion, part RAAS.
Volume depeletion, Hyperuricemia –> gout; develop in 1st 2-4 wks of therapy
Ototoxicity w/ NKCC2 isoform in cochlea

MOA of Thiazide diuretics
hydrochlorothiazide and chlorthalidone (longer acting; 2x more potent) inhibit the Na/Cl symport in the DCT
chronic use associated w/ a decrease in Ca2+ excretion (reduce kidney stones)
Thiazide use associated w/ hyperglycemia
when creatinine clearance has fallen to 40-50 mL/min, thiazides efficacy is diminished, due to reduced Na+ delivery to DCT
*Thiazides usually used in the Hypertension, nephrogenic DI

MOA of Inhibitors of Renal Epithelial Na+ Channels (K+-Sparing Diuretics)
Ameloride and Triamterene
Na+ normally reabsorbed via ENaC w/o an anion, creating a lumen negative electrical gradient –> K+/H+ secretion
drugs may be used w/ CA inhibitors, loop diuretics, thiazides
side effects make sense: hyperkalemia and metabolic acidosis
hyperkalemia caused by:
- renal failure*
- K+-sparing diuretics*
- ACEi or angII blockers*
- NSAIDs*
- dietary K+ supplements*

MOA of antagonistis of Mineralocorticoid Receptors
(Aldoesterone Antagonists, K+-Sparing Diuretics)
Spironolactone - competitively inhibit the binding of aldosterone to the MR (higher [aldosterone] –> higher effect of MR antagonists)
aldosterone binding to mineralcorticoid receptor leads to angiotensin-induced proteins (NaCl transport enhanced, the lumen-negative transepithelial voltage is increased, upregulated kinase to inhibit degradation of Na+ channels (more Na+ reabsorb)
SE: Hyperkalemia; Gynecomastia in men; loss of libido in women
*used w/ thiazide or loop diuretic in txt of hypertension and edema; dual therapy to limit hyperkalemia, enhance diuresis; reduces morbidity and death when used w/ heart failure therapy.

Edematous conditions that are treated with diuretics include:
Acute decompensated heart failure
Chronic stable heart failure
Cirrhosis with ascites
Nephrotic syndrome
Chronic kidney disease
Non-edematous condition treated with a diuretic:
The thiazides and thiazide- type diuretics are important antihypertensive drugs.
Not all generalized edema requires treatment with diuretics. When the drugs are used in non-emergent situations it is most often prudent to induce slow change rather than rapid change. Pulmonary edema is the only life-threatening edema state.
I have just given a diuretic and have a new ‘steady state’ established
What is the pathopsio that is occuring after this?
Angiotensin II, aldosterone and norepinephrine produced by diuretic-induced neurohumoral activation can all foster tubular Na⁺ reabsorption. However pharmacologic blockade of these pathways does not prevent secondary Na⁺ retention in the post-diuretic phase, i.e., when diuretic action is nil.
Now, new steady state and ECF is LOWER, but the diuretic-induced Na+ losses are offset by: Neurohumorally-mediated increases in tubular Na⁺ reabsorption at sites that are not influenced by the diuretic
Increased distal delivery of Na⁺ increases distal Na⁺ reabsorption. Furosemide administration leads to distal tubular hypertrophy and increased Na-K ATPase expression
Acute Kidney Injury
- Abrupt loss of kidney function
- Retention of urea and other nitrogenous waste products
- Dysregulation of extracellular volume and electrolytes
Creatinine
Breakdown product of creatinine phosphate in muscle
- Filtered by the kidney and used to estimate kidney function/filtration
- Inversely proportional to function
- Higher the creatinine, the lower the filtration
Blood Urea Nitrogen (BUN)
Urea nitrogen formed from protein catabolism by the liver
- Filtered by the kidney and used as an additional measure of kidney function
- High BUN generally reflects lower filtration
- Caveat: BUN can increase independent of kidney function
–Steroids, tetracycline antibiotics, or reabsorption of blood in GI tract
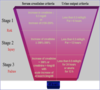
Staging System for Acute Kidney Injury
divides kidney injury into 3 stages based on absolute and change in serum creatinine and reduction in urine output.
*The higher the stage, the worse the outcome.
Issues w/ staging of Acute Kidney Injury
Based on serum creatinine and urine output (imperfect biomarkers)
By the time serum creatinine rises or urine output decreases, substantial injury may have already taken place (typically in the preceding 24-48h before serum creatinine
Causes of pre-renal acute kidney injury (2)
What does the kidney do to counteract this?
True volume depletion (loss of Na+ from ECF)
GI losses, hemorrhagic shock, renal losses, cutaneous losses
Total ECF may be increased but arterial blood volume perceived by baroreceptors in the carotid sinus and glomerular afferent arterioles is low –> edematous states
-Hepatic failure, Hepatic cirrhosis, Sepsis
decreased renal perfusion –> afferent arteriole vasodilation –> efferent arteriole constriction –>Increased filtration fraction –>higher oncotic pressure in post-glomerular capillaries –>Increase salt/water reabsorption in PCT
Activation of angiotensis II and ADH lead to increased salt and water reabsorption
(NSAIDs) block prostaglandin (blocks afferent arteriolar dilatation) and ACE inhibitors/Angiotensin receptor blockers (block efferent >afferent arteriolar constriction) prevent proper homeostasis
Diagnositic Workup of Pre-Renal Disease
pt hx
PE
Labs
–Vomiting, diarrhea, GI bleeding
–Heart failure, liver disease/cirrhosis, sepsis
PE: Orthostatic hypotension, skin tenting, dry mucous membranes
–Elevated jugular venous pressure with hypotension (heart failure), edema with hypotension
BUN:creatinine ratio >20:1
FENa: <1% (fraction of filtered sodium excreted in the urine)
Urine will have no protein, blood, or white blood cells; high specific gravity; no casts
Pre-Renal Disease Labs
BUN:creatinine ratio >20:1
FENa: <1% (fraction of filtered sodium excreted in the urine)
Urine will have no protein, blood, or white blood cells; high specific gravity; no casts
Fraction Excretion of Na+ (FENA)
What FENA would indicate prerenal disease
mesures the percent of filtered Na+ excreted in the urine
Used to differentiate b/w prerenal disease and acute tubular necrosis
•FENa is <1% in prerenal disease and indicates that the patient will be responsive to volume (i.e. IV fluids)
Discuss why tubuloglomerular feedback is important in acute tubular necrosis (ATN)
Patch necrosis of PCT and TAL –> cannot sufficiently reabsorb Na+ and Cl- –> More salt delivered to distal nephron –> macula densa senses this increase in Cl- and activates tubuloglomerular feedback, causing constriction of the afferent arteriole to reduce renal blood flow and glomerular filtration rate.
Lowering of GFR limits salt wasting and volume depletion and is a critical mechanism to prevent death in patients with ATN.
Identify the causes of acute tubular necrosis (ATN)
Ischemic, such as occurs with prolonged volume depletion, low blood pressure, or sepsis
Toxin-related, and can occur from radiocontrast media (patients who are at risk include those with underlying kidney disease, diabetes mellitus, or hypotension), drugs (aminoglycosides, amphotericin B, cisplatin), or heme pigments (result of rhabdomyolysis)
Distinguish the differences in the urinary indices between pre-renal disease and ATN (i.e. include FENa and sediment exam)
Pre-renal disease
Low sodium and chloride concentration, <10 mEq/L
BUN:creatinine ratio >20:1
FENa: <1% (fraction of filtered sodium excreted in the urine)
Urine will have no protein, blood, or white blood cells; high specific gravity; no casts
ATN
Urinary Na+ and Cl- concentration will be higher than pre-renal disease, due to dysfunction of the tubules. >20 mEq/L
BUN:creatinine is 10–15:1. Due to tubular dysfunction, not as much urea is reabsorbed into the blood, so the serum concentration will be lower than in pre-renal disease
FENa > 2%. More Na+ is excreted in the urine due to tubular dysfunction as compared to pre-renal disease in which the tubules are functioning normally.
Specific gravity of urine will be close to 1, due to loss of concentrating ability.
Low grade proteinuria due to impaired protein reabsorption in the proximal tubule
Urinary sedimentation has “muddy” brown casts; tubular cells
history, physical exam findings suggestive of acute interstitial nephritis
History of drug exposure (NSAIDs, penicillins, cephalosporins, sulfonamides, rifampin, proton pump inhibitors, ciprofloxacin), autoiummine disease (Sjogren’s syndrome, sarcoidosis) or infection (Legionella, leptospira, cytomegalovirus)
For drug-induced, onset is 3–5 days after second exposure; weeks to months after first exposure (type IV hypersensitivity reaction)
Physical Exam and laboratory evaluation suggestive of acute interstitial nephritis
Physical exam, see triad of rash, fever, and eosinophilia. Full triad only observed ~10% of the time, with fever and eosinophilia most common. This applies to drug-induced cases.
For Sjogren’s syndrome, see dry eyes, mouth
Labs
Acute rise in serum creatinine that corresponds to drug administration
Peripheral eosinophilia on drug smear
Eosinophils in the urine (>1%); not sensitive or specific Variable proteinuria
WBCs, may see WBC casts in the sedimentation
A biopsy is diagnostic
Identify 4 causes of intratubular obstruction that lead to tubulointerstial disease
Cast nephropathy: seen in multiple myeloma. Excess production of immunoglobulin light chains must be filtered into the urine and can obstruct the tubules.
Tumor lysis syndrome: after chemotherapy treatment, breakdown of large tumor causes release of uric acid and phosphate that can precipitate and block tubules.
Phosphorous-containing enemas: acute calcium phosphate deposition in the tubules, inflammation
Medications: acyclovir, sulfonamide antibiotics, methotrexate
All primarily consist of precipitation of a substance in the tubules in the setting of volume depletion and acidic urine.
Discuss the history, physical exam findings, and laboratory evaluation suggestive of cholesterol emboli (renal atheroembolic disease) and distinguish the difference in timing of serum creatinine elevation compared to radiocontrast exposure (i.e. how soon after aortic manipulation will you see a rise in creatinine compared to radiocontrast exposure)
History of aortic manipulation or coronary/renal angiography (2–8 weeks); history of atherosclerotic disease.
Causes inflammation, cholesterol emboli (ischemic injury) On physical exam, livedo reticularis rash.
Laboratory evaluation, see low serum complement, peripheral eosinophilia. Bland sediment in urinalysis.
In cholesterol emboli, the rise in serum creatinine is subacute, and occurs 2–8 weeks after the procedure. In radiocontrast exposure, serum creatinine will rise within 72 hours.
Distinguish the causes of post-renal acute kidney injury
Calculus (stone) in a solitary kidney, at the ureteropelvic junction; bilateral staghorn calculi
Anatomic abnormalities (most commonly seen in children), including ureteral strictures, stenosis, or abnormalities in ureteral valves that cause an obstruction
Benign prostatic hyperplasia (most common in men >50 years) Urethral stricture
Malignancies (extra-urinary) that compress the ureters bilaterally
Discuss the general approach to a patient with acute kidney injury
Take a good history
Were there episodes of prolonged hypotension? (septic shock)
Drug, nephrotoxin exposure: did the patient use NSAIDs, sulfonamides, contrast, enemas, aminoglycosides
Good physical exam: determine the patient’s volume status (hypovolemic or reduced effective vascular volume points to pre-renal cause); look for rash
Utrasound, to rule out obstruction
Lab values: urinalysis. Look at Na+, FENa, sedimentation, proteinuria, hematuria, crystals. Some cases warrant renal biopsy.
complications of acute kidney injury
Uremia: Symptoms from high levels of wastes in the serum. Nausea, vomiting, anorexia, dysguria (food tastes metallic), altered mental status, pericarditis
Electrolyte abnormalities
Hyperkalemia (due to decreased distal flow)
Hypokalemia (due to increased distal flow) (aminoglycosides)
Metabolic acidosis
Extracellular volume excess
Chronic kidney disease
Due to unresolved or recurrent AKI
Discuss treatment and prevention strategies that are used in acute kidney injury and identify when renal replacement therapy is warranted.
Volume resuscitate with saline or colloid (blood) if needed
Ensure renal artery perfusion during shock (use vasopressor or ionotropes)
Treat underlying infection if septic
Prevent further injury by avoiding NSAIDs, and giving sufficient water with medications known to precipitate (acyclovir)
Pretreat with sodium bicarbonate and N-acetylcysteine before giving radiocontrast agent (prevent injury)
If patient has AIN, remove offending drug; steroid if patient does not improve
Renal replacement therapy is indicated in patients who have refractory acidemia, volume overload (pulmonary edema), hyperkalemia, or uremia
Identify the 5 clinical clues that suggest the presence of secondary hypertension
Young age of onset (before third decade)
Sudden onset
Refractory or uncontrolled
Hypokalemia associated with metabolic alkalosis, without the use of diuretics
Features of an underlying cause, such as hyperglycemia in the setting of Cushing’s syndrome
What are the common causes of secondary hypertension
Renal: Renovascular hypertension, renal parenchymal hypertension
Endocrine: Primary hyperaldosteronism, Cushing’s syndrome, pheochromocytoma
Drugs: licorice (contains glycerrhetinic acid) inhibits 11–beta hydroxysteroid dehydrogenase
Identify the most common etiology of renovascular hypertension
Renovascular hypertension caused by renal artery stenosis (unilateral or bilateral)
Atherosclerosis causes 75–90% of renovascular hypertension, typically in patients >50 who have cardiovascular risk factors (tobacco use, dyslipidemia, peripheral vascular disease)
Fibromuscular dysplasia: 10–25% of renovascular hypertension (30–50 years, women>men)
Understand the relationship between renal artery stenosis and the renin-angiotensin system (pathophysiology)
- Reduced renal perfusion pressure resulting from stenosis of the arterial vasculature in one of both kidneys
- Decrease in renal perfusion pressure activates RAAS
- Angiotensin II stimulation causes the following that results in sustained hypertension:
1. Increase in sympathetic nervous system activity
2. Vasoconstriction
3. Anti-diuretic hormone release (water retention)
4. Aldosterone release from the adrenal gland (sodium retention)
Discuss the diagnostic imaging tests and possible risks associated with performing these in patients with renovascular disease and impaired renal function/GFR.
Magnetic resonance angiography: highly sensitive, specific, and noninvasive. This is the modality of choice. However, it cannot be used in patients with GFR of <30 ml/min due to the contrast agent.
CT spiral angiography with contrast: High sensitivity and specificity. Risk of nephrotoxicity in patients with impaired GFR due to the contrast agent.
Duplex doppler ultrasonography: Time consuming test with moderate sensitivity and specificity. Results are operator-dependent. However, no contrast, so safe for patients with impaired GFR. Standard for diagnosing fibromuscular dysplasia (10–25% of renovascular hypertension)
Conventional renal arteriography: Gold standard for diagnostic investigation of renal artery stenosis. Usually used pre-intervention. Risks include damage to femoral or renal artery; cholesterol embolization; contrast-induced nephrotoxicity.
Treatment of Renovascular Disease
- Treat with antihypertensives and lipid lowering therapy (if appropriate)
- Patients with fibromuscular dysplasia should try ACE inhibitor or ARB; if remain hypertensive, then can have percutanous transluminal angioplasty (PTA)
- Patients with atherosclerosis should use antihypertensives, lipid- lowering drugs, and antiplatelets. PTA is reserved for patients with uncontrolled hypertension with multiagent therapy, or rapid rise in serum creatinine with parenchymal loss.
Identify the factors involved in the pathogenesis of hypertension in renal parenchymal disease (CKD) and the most common treatment
Pathogenesis is multifactorial. Includes volume expansion via sodium and water retention, RAAS activation, activation of the sympathetic nervous system, endothelial dysfunction, and secondary hyperparathyroidism (intracellular calcium promotes constriction of vascular smooth muscle)
Most common treatment ACE inhibitor or ARB; diuretic to remove excess volume; calcium channel blocker if a third agent is needed
Know the diagnostic testing and treatment for primary hyperaldosteronism
Plasma aldosterone concentration (PAC) to plasma renin activity (PRA) ratio; if the ratio is > 30–50 and the PAC is >=15, then suggestive of primary hyperaldosteronism
*Aldosterone antagonists (spironolactone) must be discontinued for 6 weeks before testing
Confirmatory testing: sodium loading for 3 days, followed by 24 hours urine collection. Normally, aldosterone will fall with this high salt intake. If it is still high, urinary levels will be >14 ug, and indicates primary hyperaldosteronism
Can also do normal saline administered over 4 hours, followed by a plasma aldosterone level. Aldosterone should be <5 ng/dL; if >10ng/dL, confirms primary hyperaldosteronism
CT, adrenal vein sampling used to distinguish between adenoma, carcinoma, or adrenal hyperplasia
Treatment: laparoscopic removal of adenoma. If adrenal hyperplasia, treat with spironolactone indefinitely.
Identify the clinical features of Cushing’s Syndrome
Centripetal obesity: fat deposition in the face, neck, trunk, abdomen
Moon facies: fat accumulation in the cheeks, temples
Skin atrophy and abdominal striae: broad purple streaks
Acne and hirsutism: increased hair on upper lip and chin due to androgen excess
Proximal muscle weakness
Hypertension
Glucose intolerance
Discuss the diagnosis and treatment of Cushing’s syndrome/disease
Diagnosis
- 24 hour urinary cortisol excretion; positive test if concentration is 3x upper limit of normal
- Late evening salivary cortisol
- Low dose dexamethasone suppression test: dexamethasone suppresses ACTH release, leading to a suppression of cortisol secretion. A normal test is <50 nmol/L.
If two of the tests are abnormal the diagnosis is confirmed.
Treatment
Transphenoidal resection (through the nose) of the pituitary adenoma; pituitary irradiation
In the case of adrenal tumors, remove the adrenal glands.
In the case of other tumors secreting ACTH, remove the tumor
Distinguish the clinical features of pheochromocytoma
Catecholamine-secreting tumors that arise from the adrenal medulla
Triad of headache, sweating, tachycardia and hypertension.
Symptoms may be precipitated by triggers (pain, trauma, drugs, foods)
Know the diagnostic testing and treatment for pheochromocytoma
24 hours fractionated urinary metanephrines and catecholamines (NE, epinephrine, dopamine, normetanephrine); highly sensitive and specific for pheochromocytoma
Fractionated plasma metanephrines: simpler to perform test; first line. High sensitivity but low specificity (85%)
CT or MRI of the abdomen and pelvis detects most tumors; many incidental adrenal adenomas picked up, so not very specific
MIBG scinitigraphy: can scan to detect tumors if CT is negative
Treatment: Surgical removal of the tumor
Control blood pressure first
Use Alpha antagonists are first-line, like phenoxybenzamine, 7–10 days before surgery (long acting), or phentolamine, a shorter acting alpha antagonist
After HTN is controlled, use beta blockers to prevent tachycardia. If alpha blockade is not achieved, do not use beta blockers.
Can also add nifedipine (calcium channel blocker) if needed
Acute hypertensive crisis can occur during surgery; treat with parenteral nitroprusside, phentolamine, nicardipine
Understand the association between obstructive sleep apnea (OSA) and hypertension
Association of obestity, obstructive sleep apnea, and hypertension have long been established.
Apnea may contribute directly to hypertension due to abnormalities of sympathetic nervous system function and vascular reactivity
Identify the clinical features of Obstructive Sleep Apnea
How is OSA diagnosed
Daytime sleepiness, morning headache, snoring or witnessed apneic episodes, poor concentration
30–80% of patients with essential hypertension have obstructive sleep apnea
50% of patients with obstructive sleep apnea have hypertension
Polysomography (sleep study)
Discuss the treatment options available for OSA
Weight loss, avoidance of alcohol, and sedating medications (benzodiazepines, antihistamines) - depress the CNS
Continuous positive airway pressure (CPAP) splints the upper airway open
Oral appliances to hold the tongue and mandible more anterior
Surgical therapy: uvulopalatopharyngoplasty (UPPP) for those with correctable obstructing lesion
Identify the two determinants that define chronic kidney disease and discuss why one can have chronic kidney disease with a normal glomerular filtration rate (GFR)
The presence of either kidney damage or decreased kidney function for > or equal to 3 months with or without decreased glomerular filtration rate (GFR)
Kidney damage can be determined by:
Pathological: kidney biopsy
Clinically, as proteinuria (>150 mg, or >30mg albumin), glomerular hematuria (dysmorphic RBCs, RBC casts), or on imaging (polycystic kidneys, hydronephrosis, small kidneys)
*GFR can be normal and still have kidney damage pathology!

Estimating GFR with serum creatinine
how does this work
limitations
when is it most useful
Derived from the metabolism of creatine in skeletal muscle and from dietary meat intake –> released into circulation at constant rate –> stable plasma concentration
Freely filtered across the glomerulus and is neither reabsorbed nor metabolized. It is inversely proportional to GFR
*Limitations:
not accurate in patients w/ little muscle mass, dwarfism
Creatine is secreted by organic secretory pathway in PCT and certain meds (Trimetheprim-Sulfamathoxazole, Cimetadine) inhibit secretion.
Only useful when GFR is low, as creatinine changes little until GFR is <60 ml/min
Estimating GFR with Creatinine clearance
how does this work
limitations
Clearance = UV / P
U=urinary concentration of a substance, V=volume of urine per set time (in this case 24h), P=plasma concentration of a substance
*Limitations:
since creatinine is also secreted at PCT, urinary [creatinine] will be higher than was actually filtered –> creatinine clearance will excedd the true GFR by 10-20%
Also patients tend to over or under collect urine (i.e. in 24 hour sample), so hard to get an accurate clearance.
Not recommended for routine GFR assessment.
Estimating GFR with Modifications of Diet and Renal Disease (eGFR)
how does this work
limitations
when is it less accurate
Estimates GFR by incorporating known demographic and clinical variables as observed surrogates for unmeasured factors other than GFR that affect serum creatinine
Increasingly used not only to estimate GFR but follow changes in GFR
Becomes less accurate when GFR >60ml/min
Estimating serum Creatinine using Chronic Kidney Disease Epidemiology Collaboration equation
how does it work
When is it used?
Cystatin C method is not PrimeTime yet
Also estimates GFR based on age, gender, ethnicity, and creatinine
. Better accuracy than MDRD when GFR >60ml/min (may eventually replace MDRD; for now MDRD is used most often in the US)
What is the Staging in Chronic Kidney Disease (1-4)
What has Stage 3 been modified to? specific GFR intervals…what does this lead it
Stage 3 has been recently modified to 3a (GFR 46-59) and 3b (31-45) given association with higher mortality at GFR <45ml/min (particularly with cardiovascular events)

Pathophysiology of Chronic Kidney Disease
step-by-step
initial kidney insult –> nephron loss –> remaining nephrons will hyperfilter (same GFR) –> glomerular capillary hypertension –> cytokine activation and podocyte dysfunction –> proteinuria, glomerular sclerosis, tubulointerstitial fibrosis ==> Renal scarring

How does DM lead to Glomerulopathy (CKD)
DM –> mesangial expansion, thickening of GBM, glomerulosclerosis –> hyperfiltration/ glomerular capillary HTN –> Microalbuminuria (30-300mg) –> Macroalbuminuria (>300mg)
Or, the disease may progress w/o albuminuria

Describe the relationship between African-American ethnicity and progression of hypertensive-related CKD. Discuss the role of the APOL1 allele variant
Clinically what will patients also have
polymorphisms of APOL1 gene give selective biological advantage - resistance against Trympanasoma brucei rhodesiense, however, when inherited in recessive fashion –> HTN CKD with higher progression to ESRD
Stuff associated w/ longstanding HTN: retinopathy, LV hypertrophy, proteinuria (< 1g/day)
Identify the consequences of CKD and understand the relationship to cardiovascular disease
Most patients with stages 2–4 chronic kidney disease die from cardiovascular causes, and do not progress to end-stage renal disease
CKD patients have a high risk for developing cardiovascular disease, even when controlling for other shared risk factors, due to decreased GFR and increase in proteinuria
Patients will also have HTN: Na+ retention; High RAAS; High SNS; secondary hyperparathyroidism –> vasoconstrict (more Ca2+)
patients also have decreased urinary phosphorus excretion –> compensatory increase in FGF-23 (phosphatonin); increases phosphate excretion –> decrease in 1,25-VitD (FGF inhibits) –> decrease serum Ca2+–> increase in PTH –> can lead to osteomalacia, osteitis fibrosa, vascular calcification
Anemia - decreased EPO secretion from kidney
RISK FACTORS FOR CHRONIC KIDNEY DISEASE PROGRESSION TO ESRD (12)
Fat Smoking ‘Meat Head’ Males => hyperlipdemia, HTN, DM, proteinuria, hyperphosphatemia, hyperuricemia
- Proteinuria > 1.0 grams per day
- Hypertension
- Type of underlying disease (i.e. diabetes mellitus, polycystic kidney disease)
- African-American race
- Male gender
- Obesity a. Increases glomerular capillary pressure
- Hyperlipidemia a. High lipid levels are associated with a faster rate of progression
- Smoking
- Hyperphosphatemia
- Metabolic acidosis a. Bicarbonate supplementation appears to slow progression of CKD b. Mechanism thought to be due to reduction in tubulointerstitial inflammation
- High protein diet a. Increases glomerular capillary pressure
- Hyperuricemia a. Data emerging that treatment to reduce uric acid levels may slow the rate of GFR loss
INTERVENTIONS THAT SLOW PROGRESSION OF CHRONIC KIDNEY DISEASE
*The most important intervention is adequate BP control! (<130/80) (140<90 in African Americans)
Renin-angiotensin-aldosterone antagonism: ARBs, ACEIs
Control phosphorus levels: reduce intake or take binders
Treat metabolic acidosis: sodium bicarbonate
Stop smoking
Correct anemia: (EPO)
Use a Statin
Low protein diet
Treat underlying disease - make sure blood sugar control is tight
Which form of renal replacement therapy confers the best survival advantage
pre-emptive transplant (transplant prior to dialysis)
What happens physiologically when the kidney must excrete out greater amounts of acid
inscreases ammonia production (derived from metabolism of glutamine) with a resultant increase in NH4+ (ammonium) in the urine
How is urine propelled through the upper urinary tract and what happens when urine propulsion is blocked?
Peristaltic contractions propel the urine towards the bladder (2–6 cm/s). Pacemaker cells initiate the spontaneous action potentials that lead to contractions. The fastest pacemaker cells are located in the calyces. Urine enters the bladder at the ureterovesical junction; since pressure in the bladder is normally low, it fills easily.
*If the ureter is blocked (by a stone, for example) or if the bladder pressure is high (neurologic disease), then the ureter becomes distended and hydronephrosis occurs.
If unilateral, the other kidney can compensate.
If bilateral, can cause kidney failure or irreversible kidney damage. Very serious.
Describe the functional anatomy of the human bladder and urethra
The bladder is composed of diffuse smooth muscle fibers. It contains a body and a base; the body is distensible to allow filling with urine, while the base is fixed, and is where urine enters and exits.
The bladder base contains the trigone and bladder neck (point which connects to the urethra)
The bladder outlet includes structures involved in continence and voiding: bladder base, urethra, external sphincter
The bladder neck and proximal urethra are the internal sphincter, which does contribute to continence

What are the essential differences between the male and female urethra with regards to the mechanisms contributing to urinary continence
Female
Female:
The urethra is fused to the anterior wall of the vagina. It is short in females.
Primary zone of continence is the external sphincter, located at the urogenital diaphragm. It is augmented by the pelvic floor muscles.
Continence is promoted by the tone of the external sphincter; the support of the anterior wall of the vagina; and the coaptation of the anterior and posterior walls of the urethral lumen (sticking together).
Childbirth, nerve damage, loss of estrogen all impact continence in women. Urinary incontinence is more common in women.

What are the essential differences between the male and female urethra with regards to the mechanisms contributing to urinary continence
Male
Male:
Primary zone of continence is the external sphincter, located at the membranous urethra. It is augmented by the pelvic floor muscles.
There is a secondary zone of continence located at the bladder neck/prostatic urethra that closes during ejaculation and contributes to continence.
Areas of the long male urethra are the: prostatic urethra; membranous urethra; bulbar urethra (penile base); and the pendulous urethra (penile shaft).

Understand and describe the autonomic physiology of the lower urinary tract and the neural mechanisms and physiologic events involved in urine storage and micturition.
The detrusor muscle is rich in muscarinic cholinergic receptors; activation of these M3 receptors results in contraction of the detrusor (parasympathetic activation = elimination mode)
During contraction of the detrusor, it is thought that a second system using nitric oxide is activated to relax the bladder base/outlet and allow for elimination
The dome of the bladder (body) has many beta adrenergic receptors; activation of these Beta-3 receptors results in relaxation of the detrusor (sympathetic activation = storage mode)
The base of the bladder and proximal urethra have many alpha adrenergic receptors; activation of these Alpha-1 receptors (NE) results in contraction of the bladder neck, promoting storage of urine (sympathetic activation = storage mode).
Males have more Alpha-1 receptors in the bladder neck to prevent retrograde ejaculation into the bladder during ejaculation.
Phase 1 of the micturition cycle: Storage
steps
The detrusor must be relaxed, while the outlet and external sphincter must be contracted.
The bladder is normally very compliant; it fills at low pressure and can stretch a great deal (compliance is volume change over pressure change)
There are afferents that detect the amount of stretch in the bladder wall; this information is sent to higher centers and affects the decision to void
People with stiff/ non-compliant bladders will be signaled to void at lower volumes.
In storage mode, the cortex inhibits the PMC (pontine micturition center). As the bladder fills, afferent fibers are fired, activating a reflex in the lumbar and sacral spinal cord. This reflex causes an outflow of sacral somatic activity via the pudendal nerve that contracts the external sphincter, and sympathetic activity that:
Relaxes the detrusor via beta 3 receptors
Contracts the muscle of the bladder neck and urethra via alpha 1 receptors
Inhibits parasympathetic transmission from the ganglia in the wall of the bladder
phase of the micturition cycle: Emptying
steps involved
The detrusor muscle contracts, and the outlet relaxes.
The cortex decides to void, releasing inhibition of the PMC (pontine micturition center). This sends signals to the lumbar (sympathetic) and sacral (parasympathetic) cord to do the following:
Inhibit the pudendal nerve, allowing the external sphincter to relax
Inhibit the hypogastric nerve lumbar sympathetics, which aborts the actions discussed above, i.e. aborts the guarding reflex
Stimulates parasympathetic outflow from the sacral micturition center, causing contraction of the detrusor and relaxation of the bladder neck and proximal urethra
The PMC can make the decision to void in the absence of cortical input
Describe the cellular/molecular events leading to bladder contraction
Acetylcholine activates muscarinic M3 receptors on the detrusor. This inhibits the neurons in the parasympathetic ganglion from firing, as well as activates a G-protein coupled mechanism that activates phospholipase C (PLC). PLC activation results in the generation of IP3. This leads to opening of calcium channels in the cell membrane and in the sarcoplasmic reticulum, causing increased contraction of the smooth muscle.
Calcium binds to calmodulin, which then activates myosin light chain kinase. MLCK phosphorylates the myosin light chain, and contraction occurs (via cross-bridging to actin).
ATP is hydrolyzed slowly, and the force generated lasts for a relatively long period of time.
Describe the cellular/molecular events leading to bladder relaxation
Adrenergic activation of beta-3 receptors in the detrusor causes activation of adenylyl cyclase, which increases cAMP in the cell. Increased cAMP prevents phosphorylation of the myosin light chain, preventing cross-bridging and thus leading to relaxation of the muscle.

Detrusor overactivity: steps behind this
symptoms associated
the detrusor contracts involuntarily, causing a sudden need to urinate (urge incontinence). The patients may not be able to make it to the bathroom when this urge comes on. Characterized by urinary urge and urinary frequency. Failure to store due to the bladder (Wein)

Urethral Incompetence:
associated symptoms
abnormal function of the bladder outlet. May be a problem with the bladder neck or with the urethra. Patients report stress incontinence (urination with coughing, laughing, etc). In women, it occurs after menopause or after vaginal childbirth. In men, it typically occurs after prostate surgery due to damage to the pedundal nerve. Failure to store due to the outlet (Wein).

Overflow incontinence:
associated symptoms
the bladder is unable to empty, either due to an obstruction or a failure of the detrusor to contract. As a result, the bladder becomes completely distended with urine, and the urethra continuously leaks. Failure of the bladder to empty (Wein)

Mixed incontinence:
associated symptoms
can occur, and clinically this often presents as stress incontinence and urge incontinence together.

according to Wein classification, Failure to store urine can be from (3)
Due to a problem of the outlet
Due to a problem of the bladder
Due to a problem of both bladder and outlet (mixed)
according to Wein classification, Failure to empty urine can be from (3)
Due to a problem of the outlet
Due to a problem of the bladder
Due to a problem of both bladder and outlet (mixed)
What are the physiologic characteristics of bladder function that can be learned through urodynamic testing of the bladder
Bladder capacity and compliance Bladder sensation
The detrusor pressure at which the bladder stores and empties urine; the urinary flow rate produced by the voiding detrusor pressure
Determine if the bladder is obstructed by examining the pressure-flow relationship during micturition
Determine if the bladder exhibits detrusor overactivity, which can lead to urinary incontinence
Determine the presence of a weak urinary sphincter and the intravesical pressure at which incontinence occurs due to the weak sphincter
Describe the common expected patterns of voiding dysfunction encountered with neurologic injuries at different levels of the central nervous system.
above the brainstem
For lesions above the brainstem, patients have detrusor overactivity. This occurs because there is less inhibition on the PMC, causing the PMC to initiate voiding more frequently. Urinary frequency, urgency, nocturia, urge incontinence.
Describe the common expected patterns of voiding dysfunction encountered with neurologic injuries at different levels of the central nervous system.
AT the brainstem
For lesions in the brainstem, the patient will have complex and variable voiding dysfunction (if they survive)
Describe the common expected patterns of voiding dysfunction encountered with neurologic injuries at different levels of the central nervous system.
immediately after spinal injury
Immediately after spinal cord injury, regardless of the level, there is a period of spinal cord shock in which there is decreased excitability at and below the lesion. Usually this causes an areflexic, non-contractile bladder with a closed bladder outlet. The bladder fails to empty due to both the bladder and the outler (overflow incontinence).
Describe the common expected patterns of voiding dysfunction encountered with neurologic injuries at different levels of the central nervous system.
at lesions above S2
For lesions above S2 (thoracic spinal injury): the patients typically have detrusor sphincter dyssynergia. Voiding is not coordinated due to lack of input from the PMC. The external sphincter cannot relax due to lack of input from the PMC, and tends to contract as the detrusor contracts. Spinal reflexes attempt to empty the bladder, but cannot due to contraction of the external sphincter; the detrusor is hyperactive and the bladder does not empty completely. Failure to empty due to the outlet, and failure to store due to the bladder.
Describe the common expected patterns of voiding dysfunction encountered with neurologic injuries at different levels of the central nervous system.
lesions below S2
For lesions below S2 (upper lumbar or lower thoracic injury): the patients typically have detrusor areflexia and a fixed external sphincter. The bladder fails to contract and the external sphincter remains contracted due to lack of voluntary control. Urinary retention with overflow incontinence (failure to empty due to the bladder).
Describe the symptoms, common causes, and the bladder physiology involved in both the early and late stages of obstructive uropathy
Obstructive uropathy causes weak and intermittent urinary stream; urinary hesitancy; straining to void; sensation of incomplete emptying.
In early obstructive uropathy, the detrusor will thicken to compensate for the obstruction. This causes the detrusor to become overactive, leading to urinary urgency and urge incontinence. Urinary frequency increases, because the bladder does not empty completely and the functional capacity is reduced.
With later stages, the bladder decompensates and can no longer empty. Post-void residuals increase. Untreated, it will progress to urinary retention with hydronephrosis, which can progress to renal failure in some cases.
Difference b/w volatile and non-volatile acid production
volatile comes from CO2 (excreted from the lungs) = 24,000 mMol produced per day from normal metabolism
non-volatile: derived from the metabolism of certain amino acids, phosphorus-containing compounds, and organic acids. They are excreted by the kidneys. 50–100mEq is produced per day by normal metabolism.
- Excretes acids by combining ions with urinary buffers to form titratable acid or with ammonia to form ammonium
- Reabsorbs bicarbonate
normal values for pH, pCO2, HCO3-

diagram normal acid-base homeostasis
If respiratory acidosis develops, the kidneys will compensate by increasing H+ secretion and increasing bicarbonate (via ammoniagenesis, which increases the amount of ammonium in the urine)
If respiratory alkalosis develops, the kidneys will compensate by increasing bicarbonate secretion
Acid produced by normal metabolism is excreted by the lung (CO2) and the kidneys (nonvolatile acids) to maintain acid/base balance

Henderson-Hasselbach equation
It is used to calculate pH based on the ratio of bicarbonate to carbon dioxide tension (i.e. the arterial blood gas partial pressure of carbon dioxide)

Metabolic acidosis
characterized by a decrease in pH as well as a decrease in bicarbonate
Metabolic alkalosis
characterized by an increase in pH as well as an increase in bicarbonate.
Respiratory acidosis
characterized by a decrease in pH, and an increase in dissolved carbon dioxide (pCO2)
Respiratory alkalosis
characterized by an increase in pH and a decrease in dissolved carbon dioxide (pCO2)
How to compensate Metabolic Acidosis
Metabolic acidosis: to compensate for the decrease in pH, ventilation increases in an attempt to reduce pCO2 and raise the pH. This does not raise the pH all the way to normal but does raise it towards normal.
How to compensate Metabolic Alkalosis
Metabolic alkalosis: the respiratory compensatory response is variable and takes hours to develop. The response is hypoventilation in an attempt to raise pCO2 and lower the pH. The pCO2 can only rise to about 55–60 mmHg. Often patients have other disease states and symptoms that cause hyperventilation, thus counteracting the compensatory response
How to compensate for Respiratory Acidosis
Respiratory acidosis: acute response is increase in plasma bicarbonate (from non-carbonic acid buffers). Chronic (~72 hours) response is an increased acid excretion from the kidneys due to increased ammoniagenesis, with bicarbonate retention
How to compensate for Respiratory alkalosis
Respiratory alkalosis: acute response is due to H+ released from non carbonic acid buffers. Delayed response in the kidneys of increased HCO3– excretion, reduced excretion of ammonium and titratable acid. In chronic cases, the pH can return to within normal range.
What is the anion gap
what is normal range
what blood protein can affect this
Total cations = total anions in blood (no net charge)
The anion gap is calculated by Na+ — (HCO3– + Cl-)
The normal range for anion gap is 8–12 mEq/L
Albumin can affect the anion gap. The anion gap falls 2.5 mEq/L for each 1 g/dL reduction in albumin below the normal range. Corrected AG = AG + 2.5(4–measured albumin)

Describe the major causes of elevated anion gap metabolic acidosis and normal anion gap (hyperchloremic) acidosis
Elevated anion gap metabolic acidosis occurs due to a gain of nonvolatile acid, while normal anion gap acidosis occurs due to a loss of base.

how does the kidney compensate for respiratory acidosis and alkalosis
In respiratory acidosis, the kidneys reduce or eliminate HCO3 excretion in the urine; in addition, they increase titratable acids and ammonium excretion. This increases the net acid excretion in an attempt to raise the pH.
In respiratory alkalosis, the kidneys at first release H+ from non-carbonic acid buffers; later, there is an increase in excretion of bicarbonate, and reduced excretion of ammonium and titratable acid.
pathogenesis of metabolic alkalosis
Metabolic alkalosis is a primary disorder that causes the plasma bicarbonate (HCO3–) to rise. The pathogenesis of this disorder requires both the generation and maintenance of this process.
Decreased renal excretion of HCO3– (often related to Cl- depletion or deficiency)
Causes include GI H+ loss (vomiting, NG suction), renal H+ loss due to diuretics (loop and thiazides), primary mineralocorticoid excess, Bartter’s, Gitelman’s, & Liddle’s Syndromes, or alkali administration

Describe the clinical approach to acid-base disorders
History and physical examination
Check the pH. Is the patient acidemic or alkalemic?
Look at pCO2 and HCO3–. Is the disorder respiratory or metabolic?
Is there appropriate compensation?
Is there an elevated anion gap? (This may be the only clue to a mixed acid-base disturbance)
3 modalities of Renal Biopsy Examination
Light Microscopy (Basic morphology)
Immunofluorescence Microscopy (Immune deposits)
Electron Microscopy (Ultrastructure)
*all samples must have cortex and glomeruli
Nephrotic syndrome consists of what 4 factors that lead to renal biopsy
Nephrotic syndrome: dysfunction of glomerular podocyte Proteinuria (>3.5gm/24hrs)
Hypoalbuminemia - pitting edema
Hypogammaglobulinemia - increased risk of infection
Hyperlipidemia/ lipiduria - may result in fatty casts in urine
Hypercoagulable state - due to loss of antithrombin III
Acute nephritic syndrome is associated with what 4 factors leading to renal biopsy
What can acute nephritic syndrome eventually lead to?
Clinical condition associated w/ glomerular capillary dysfunction/inflammation (active glomerulonephritis)
Hematuria
Proteinuria
increased BP
Edema
increased serum creatinine
Active urinary sediment - Red blood cells and casts (made of red blood cells, white blood cells and/or epithelial cells) detected in the urine; indicates active glomerulonephritis
Rapidly progressive glomerulonephritis - severe form of acute nephritic syndrome w/ a rapid rise of serum creatinine (renal emergency)
Persistent asymptomatic urine abnormality
how does it lead to renal biopsy
Usually subnephrotic range proteinuria (< 3.5 gm/24 hours) or persistent/recurrent microscopic hematuria.
How does Renal Failure lead to a renal biopsy
- Elevated serum creatinine
Acute - Glomerular, vascular or tubulointerstitial disease
Chronic - Advanced renal disease, usually irreversible, due to a variety of primary diseases.
Identify the 4 clinical findings in nephrotic syndrome and understand how these findings develop (pathophysiology of nephrotic syndrome)
Overall dysfunction of podocyte
Glomerular proteinuria: abnormalities in the podocytes allows for increased filtration of macromolecules across the glomerular capillary wall. Albumin is the principal urinary protein.
Hypoalbuminemia: consequence of urinary albumin losses. Hepatic synthesis of albumin increases, but it cannot replete the serum levels.
Edema: Due to hypoalbuminemia, plasma oncotic pressure is decreased and fluid moves into the interstitial space. Decreased effective vascular volume causes activation of RAAS, and leads to sodium and water retention (aldosterone) and reduced ANP release.
Hyperlipidemia, lipiduria: Decreased oncotic pressure stimulates the liver to produce lipoproteins, which manifests as hypercholesterolemia, hypertriglyceridemia. The lipid in the urine becomes trapped in protein material in the tubules; lipid casts. Oval fat bodies (enclosed by cytoplasmic membrane of degenerated cells)
Complications of nephrotic syndrome
Altered Coagulation –> Thromboembolism
Loss of anticoagulant factors thru glomerular capillaries (antithrombin II) into urine leads to increased pro-coagulant production in the liver.
Volume depletion from diuretic therapy + reduced oncotic pressure in the vasculature leads to concentration of blood, and increased platelet aggregation.
Renal vein thrombosis Infection - loss of fluid across glomerulus, hemoconcentration in post-glomerular circulation
*Infection Due to immunoglobulin loss in the urine
Staphylococcus, Streptococcus pneumoniae are common
Generalized treatment of Nephrotic Syndrome
ACEi or ARB (reduce intraglomerular pressure, reduce proteinuria)
Loop diuretics, low Sodium diet
Strict BP control (<130/80)
Statin to treat hyperlipidemia
Differences b/w Primary Glomerular Diseases
vs.
Systemic Diseases
That cause nephrotic syndrome
Primary Gomerular Diseases: “immune podocytopathies”
a. Membranous nephropathy
b. Minimal change disease
c. Primary focal segmental glomerulosclerosis (FSGS)
d. Idiopathic/autoimmune membranoproliferative glomerulonephritis
Secondary - Systemic Diseases
a. Diabetic nephropathy
b. Amyloidosis
c. Systemic lupus erythematosis
Minimal Change Disease
Epidemiology
Etiology
Pathogenesis
Where would you see change on biopsy?
Clinical Course of Treatment
Most common cause of nephrotic syndrome in children (90% <5 years, 50% >10 years)
Etiology: idiopathic, NSAIDs, or neoplasm (Hodgkin’s disease, lymphoma, leukemia
Pathogenesis: Systemic T cell dysfunction produces a glomerular permeability factor (cytokines) that injures podocytes. Foot processes flatten and merge, causing selective proteinuria (loss of albumin, but not immunoglobulin)
On light microscopy, not much evident (“minimal change”). Immunofluorescence is negative. On EM, see diffuse effacement of podocyte foot processes.
Nonspecific Diuretic/Low Na+; High Dose Sterioids (prednisone); Cyclophosphamide (steroid dependent); Cyclosporine (steroid resistant patients)

Primary Focal Segmental Glomerulosclerosis
Epidemiology
Etiology
pathogenesis
clinical course
treatment
Etiology: Idiopathic, Genetic/familial (involves genes encoding slit diaphragm proteins) *Most common in Hispanics/ African Americans
Pathogenesis is systemic T-cell dysfunction (as in MCD; on the same disease continuum, but FSGS is less responsive to treatment)
Presence of a circulating toxin (supported by rapidity of relapse after transplant)
On light microscopy, see focal glomerular sclerosis of a segment of the glomerular capillary tuft.
Immunofluorescence: Trapping of C3 and/or IgM in areas of sclerosis.
Electron: Diffuse effacement of podocyte foot processes
*May present with acute or insidious onset –> ESRD (if untreated)
Treatment: – Nonspecific therapy (ACEi/ARB/loop/lowNa+/BP control/statin for hyperlipidemia); High Dose steroids (prednisone); if no response, then may have familial FSGS

Secondary Focal Segmental Glomerulosclerosis
Epidemiology
Etiology
pathogenesis
clinical course
What would you see on biopsy?
treatment
Secondary etiology occurs in many forms of renal injury and systemic disease: loss of renal mass, obesity, HIV, sickle cell disease, drugs (pamidronate, heroin)
Pathogenesis related to hyperfiltration + increased glomerular capillary hypertension that causes podocyte injury; or direct injury to the podocyte from a virus (HIV)
Presents with nephrotic range proteinuria, rather than the full nephrotic syndrome (absence of hypoalbuminemia, lipid abnormalities)
On light microscopy, see focal glomerular sclerosis of a segment of the glomerular capillary tuft.
On immunofluorescence, no specific immune complex deposition
On EM, see diffuse podocyte foot process effacement in primary FSGS; variable effacement in secondary FSGS
onset is always insidious
If untreated then leads to ESRD

3 proposed mechanisms for the pathogenesis of immune-complex mediated renal disease
Antigen-antibody complexes form in the blood and then become trapped in the renal tissue.
Circulating antigen deposits in the kidney, then the antibody binds to it.
Renal proteins act as auto antigens, and then antibodies recognize and bind to them.
Membranous nephropathy
epidemiology
etiology
pathogenesis
clinical course of treatment
most common nephrotic syndrome in Caucasian adults
Primary/idiopathic (70%); Secondary (30%): Hep B and C, Malignancy, SLE, drugs (NSAIDs, penicillamine)
Pathogenesis: Primary - Autoimmune disease w/ IgG antibodies directed against Type-M phospholipase A2 receptor on podocyte foot process.
Secondary: caused by circulating IgG antibodies against extrinsic antigens from viral proteins, tumor proteins, drug-related substances.
Immune complex activates complement cascade, causing assembly of the membrane-attack complex (MAC) which inserts into the podocyte membrane, causing podocyte injury –> proteinuria (podocyte foot process effacement; loss of podocytes from apoptosis and necrosis.
Light Microscopy: Thick GBM w/ “spike and dome” appearance
Immuno/Electro: subepithelial granular deposits
Spontaneous remission occurs in up to 30% of patients at 5 years. Progresses to ESRD in 40% at 15 years if not treated.
Nonspecific therapy: ACEi/ARB, loop diuretic, low Na+, BP control, statin for hyperlipidemia.
If persists: Cyclophosphamide + prednisone x 6 months

How does diabetic nephropathy occur
biopsy findings
treatment
most common cause of ESRD in the US
Hyperglycemia leads to nonenzymatic glycosylation of vascular basement membrane resulting in hyaline ateriolosclerosis. Glomerular Efferent arteriole more affected leading to High glomerular filtration pressure –> Microalbuminuria
–> nephrotic syndrome: Sclerosis of the mesangium with formation of Kimmelsteil-Wilson nodules
ACEi/ARB reduce glomerular capillary pressure; Strict blood sugar control

Distinguish 2 types of amyloid in renal disease and identify the treatment for each
caused by extracellular deposition of abnormally folded proteins that deposit in the mesangium, resulting in nephrotic syndrome.
AL type (primary)
- Multiple myeloma; amyloid made of monoclonal immunoglobulin light chains
AA type (systemic reactive)
-chronic inflammatory disease (RA, inflammatory bowel, hepatitis); amyloid made of serum amyloid A protein
Characterized by apple-green birefringence under polarized light after staining with Congo red; haphazardly arranged fibrils
Treatment: Nonspecific ACEi/ARB/loop/low Na+diet /BPcontrol/hyperlipidemia
Primary: Chemotherapy, Peripheral stem cell transplant
Secondary: Treat underlying disease
Cystitis
what does it normally present as
what organisms cause it?
Urinalysis
Dipstick
Culture
infection of the bladder can lead to *SCC
Presents as dysuria, urinary frequency, urgency, suprapubic pain, systemic signs are usually absent (fever, chills)
E coli (80%) >>> Proteus, Klebsiella, Enterobacter, Schistosoma (middle eastern countries)
Urinalysis - cloudy urine w/ > 10 WBCs/ high power field
Dipstick - Positive Leukocyte esterase (due to pyuria) and nitrites (bacteria convert nitrates to nitrites)
Culture - greater than 100,000 colony forming units
Acute cystitis
Gross
Microscopic

Gross: Hyperemia of mucosa, gray-tan exudate
Hemorrhage
Microscopic: Neutrophilic infiltrate, hemmorrhage, ulceration of mucosa

Chronic Cystitis
gross
micro
Hyperemia of mucosa
Chronic infiltrate, mostly lymphocytes, plasma cells; reactive-appearing epithelium; Fibrosis and thickening of muscularis propria (decreased contractility)

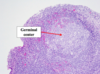
Chronic Follicular Cystitis
- aggregation of lymphocytic infiltrate with lymphoid follicles

Eosinophilic Cystitis
-submucosal eosinophilic infiltrate, fibrosis and occasional giant cells due to allergies, parasites, or idiopathic

Malacoplakia
unique form of chronic cystitis; caused by E Coli infection
immunosuppresed patients (esp. transplant)
Gross: multiple yellowish plaques in mucosa and submucosa
Micro: Large foamy macrophages, multinucleate giant cells, lymphocytes, Michaelis-Gutmann bodies (round, intracytoplasmic concretions, thought to represent defects in phagocytosis)


Polypoid cystitis
inflammation resulting from irritation to bladder mucosa (commonly indwelling catheter)
Broad, polypoid mucosal projections due to submucosal edema
Can be confused w/ papillary urothelial carcinoma

Emphysematous cystitis
-Inflammation associated with formation of air-filled spaces

Cystitis cystica
- Nests of transitional epithelium gown downward into lamina propria; may have central cystic spaces
- Can be seen in normal bladders, but also in the setting of inflammation and metaplasia
Interstitial Cystitis
more prevalent in men or women?
what does it lead to
symptoms
most frequent in women
- Inflammation and fibrosis in all layers of the bladder wall, often with ulceration – Can mimic gross appearance of carcinoma in situ (CIS)
- Highly incapacitating and difficult to treat
- suprapubic pain, frequency, urgency, hematuria, dysuria
Acute pyelonephritis caused by what pathogens?
caused by ascending infection from bladder from gram negative rods E coli, Klebsiella, Enterobacteria
Predisposing factors leading to Acute Pyelonephritis
Urinary tract obstruction w/ urine stasis
Instrumentation of urinary tract (including bladder catheters)
Vesicoureteral reflux
Pregnancy (due to higher incidence of bacteriuria, UTI, and thus pyelonephritis)
Renal Disease
Diabetes Mellitus
Immunosuppresion
Acute Pyelonephritis pathophysiology
Colonization of urethra then bladder (usually intestinal flora)
Bacterial adhesion molecules interact with receptors on the urothelium for adhesion (P- fimbriae) –> promote migration
Multiplication of organisms in bladder from outflow obstruction, bladder dysfunction –> STASIS –> bacterial growth
The organism moves to the upper tract, either due to instrumentation/catheterization, or due to Vesicoureteral reflux from the bladder back up the ureters (occurs during micturition)
As urine refluxes, it moves from the renal pelvis to the medulla (intrarenal reflux), and can move back into collecting ducts at the compound papillae of the upper/lower kindey poles (they have a divet in the middle that allows this to occur)

Major signs and symptoms of acute pyelonephritis and describe the major laboratory features of acute pyelonephritis
Fever, flank pain, WBC casts, generalized constitutional symptoms of cystitis (urgency, frequency dysuria)
Laboratory findings:
leukocytosis with left shift;
pyuria (leukocytes in urine);
leukocyte casts in urine (upper urinary tract disease);
positive urine culture; positive blood cultures in 15–30% of patients (diabetics, elderly, immunosuppressed)

acute pyelonephritis
Focal abscesses or confluent, wedge-shaped areas of suppuration
Patchy neutrophilic infiltrate in interstitium and within tubules; abscess formation with destruction of engulfed tubules


acute pyelonephritis
Patchy neutrophilic infiltrate in interstitium and within tubules; abscess formation with destruction of engulfed tubules

What are the common consequences/complications of acute pyelonephritis
most cases recover with antibiotics, usually never progress to disease, but bacteria may still be present in urine (culture)
Complications: Bacteremia/sepsis - 15-30%
Papillary necrosis - usually seen in diabetics
Pyonephrosis - exudate fills renal pelvis and ureters, seen in severe obstruction
Perinephric abscess - inflammation spreads into perinephric fat
Polyomavirus
when is usually seen
what will you seen microscopically
Important viral cause of acute pyelonephritis in renal allografts
immunosuppresion leads to reactivation of latent virus w/ infection of renal tubular epithelial cells, resulting in tublointerstitial inflammation (intranuclear viral inclusionsb in tubular cells)

Pathophysiology of Chronic Pyelonephritis

interstitial fibrosis and atrophy of tubules due to multiple bouts of acute pyelonephritis
due to vesicoureteral reflux (children) or chronic obstruction (BPH or cervical carcinoma)
Leads to cortical scarring with blunted calyces; scarring at upper and lower poles - vesicoureteral reflux

Common drugs associated with allergic interstitial nephritis
is a hypersensitivity reaction
Penicillins
NSAIDs
antibiotics - cephalosporins, sulfonamides
diuretics - thiazides
acetaminophen (rare)
Describe the clinical presentation, treatment of allergic interstitial nephritis and understand potential consequences of untreated allergic interstitial nephritis
Fever, Skin rash, Eosinophilia, acute renal insufficiency - 50% patients, abnormal liver chemistries, Eosinophiluria
Not dose dependent w/ drug use; onset usually after 2 weeks on drug
Treat by discontinuing the causative drug; put on steroids; supportive care
Continuing the drug may cause permanent injury to the kidney due to scarring

Know the pathophysiologic mechanisms underlying the development of light chain cast nephropathy (myeloma kidney) and describe the histologic appearance in tissue

Abnormal synthesis of large amounts of immunoglobulin light chains (Bence Jones protein) by the neoplastic plasma cells
Light chains directly toxic to renal tubular cell epithelium
Light chains combine with Tamm-Horsfall protein and form obstructive casts within the tubules
tubular cell injury causes reactive inflammatory response and edema –> can lead to chronic injury (fibrosis)
Histology: large casts in tubular lumen –> Giant cell reactions –>plasma cells, lymphocytes. Late = interstitial fibrosis

How can BUN increase independent of kidney function
Steroids, tetracycline antibiotics, or reabsorption of blood in GI tract
5 principles of Nephritic Syndrome
- Glomerular disorders characterized by glomerular inflammation and bleeding.*
1. Limited Proteinuria (150mg - 3.5 g/day) MPGN/Lupus Nephritis >3.5
2. Oliguria (< 500 ml urine per day)
3. Azotemia (BUN:Creatinine ratio > 15)
4. HTN due to Salt retention –> periorbital edema
5. Hematuria of glomerular orgin: RBC casts and dysmorphic RBCs in urine
nephrotic syndromes with nephritic features (3)
Membranoproliferative Glomerulonephritis: Thick glomerular BM w/ ‘tram-track’ appearance due to immune complex deposition (granular IF); Renal biopsy is definitive
Type 1: subendothelial (associated w/ HBV and HCV)
Type 2: dense deposit disease; associated w/ C3 nephritic factor (autoantibody that stabalizes C3 convertase, leading to overactivation of complement, inflammation, and low levels of circulating C3); poor response to steroids; progresses to renal failure
Lupus nephritis

Membranoproliferative glomerulonephritis
etiology
pathogenesis
course
treatment
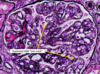
Heaptitis C (HCV, HBV, chronic bacterial infections (subacute bacterial endocarditis, ventriculoatrial shunt infection.
Neoplasia: Multiple myeloma, chronic lymphocytic leukemia
Pathogenesis: immune complex deposition –> activate complement (classical pathway) –> C3/C4 low in serum; renal biopsy definitive
biopsy: duplication of GBM ‘tram-track’ appearance; granular immune deposits IgG and/or IgM and C3 (IF); subendothelial deposits
Prolonged course w/ slow rates of disease progression; Poor response to steroids; Progress to chronic renal failure

C3 glomerulopathies: Dense Deposit Disease; C3 Glomerulonephritis
etiology
pathogenesis
course
treatment

DDD: overactivation of complement, inflammation, and low levels of circulating C3; associated w/ “Drusen” in Bruch’s membrane of retina, and partial lipodystrophy
Pathogenesis: C3 nephritic factor (autoantibody that stabalizes C3 convertase, leading to overactivation of complement); deficient/mutated regulatory proteins: Factor H, Factor I. Results in persistent degradation
Course: DDD has poor prognosis (50% progressing to ESRD in 10 years). It is common for the disease to recur even after transplantation. Prognosis and progression with C3 glomerulonephritis is unknown.
Treatment: Nonspecific therapy as with nephrotic syndrome. Can be treated with plasma exchange for patients with C3 nephritic factor (replace with frozen plasma or albumin to get rid of the factor causing complement activation). For patients with regulatory deficiency, can infuse with plasma (do not need to replace). New drug Eculizumab: monoclonal antibody to complement protein C5, preventing formation of MAC; very expensive drug, off label use.

Compare the serum complement abnormalities in immunoglobulin-mediated MPGN and Dense Deposit Disease.
MPGN: due to systemic complement activation, serum C3 and C4 are both low as well as total complement.
DDD: due to alternative complement activation, only serum C3 and total complement is low. Assay for C3 nephritic factor, Factor H, Factor I
Compare the characteristic light (LM), immunofluorescence (IF) and electron microscopy (EM) findings of immunoglobulin-mediated:
MPGN
Dense Deposit Disease
C3 glomerulonephritis
MPGN: LM: hypercellular, lobulated glomeruli; Duplication of GBM (‘tram-tracking)
IF: IgG, IgM, or C3 subendothelial deposits along GBM/mesangium
EM: mesangial/subendothelial immune deposits; Duplication of GBM
DDD: LM:similar to MPGN, duplication of GBM less prominent
IF: mesangial/capillary wall staining w/ C3
EM: Linear deposition of electron dense material along GBM ‘ribbon-like’
C3 glomerulonephritis: Variable patterns of mesangial expansion and cell proliferation on light microscopy. Variable patterns of immune complex deposition on EM; appearance is not as dark in the GBM
IgA Nephropathy
epidemiology
etiology
pathogenesis
clinical presentation
treatment

Most common glomerulonephritis world-wide (highest asian/causcasian populations)
Environemental Ag’s trigger mucosal immune response that forms pathogenic IgA immune complexes. Antigen may be viral (follow upper resp/GI infections), Bacterial, Food.
Pathogenesis: Formation of IgA immune complexes that are abnormally glycosylated and cannot be cleared by the spleen. The complexes are recognized as abnormal by the immune system and a second antibody, IgG, is formed against it (they recognize the abnormal hinge region where the glycosylation occurs). These large macromolecules then deposit in the mesangium of the kidney. Complement is activated.
Diagnosis: Renal biopsy. Serum complement is normal, as the activation occurs locally and not systemically.
Clinical presentation: ~40% have gross hematuria following URI (synpharyngitic hematuria). ~30% have microscopic hematuria and proteinuria discovered incidentally w/ RBC casts. 5–10% present with acute nephritic syndrome, and some present with rapidly progressing glomerulonephritis (RPGN). May be associated with skin or GI abnormalities; dermatitis herpetiformis, celiac sprue, cirrhosis, IgA vasculitis - Henoch-Schonlein Purpura
Treatment: Prognosis is variable, with 10–40% progressing to chronic renal failure. If renal function is normal and proteinuria is <500 mg, then no treatment. If proteinuria > 1g, then treat with ACE inhibitors or ARBs. Fish oil. Steroids for patients with proteinuria >1g or with progressive disease.

Post-Infectious Glomerulonephritis
epidemiology
etiology
pathogenesis
clinical presentation
treatment

Epidemiology: Most common cause of acute nephritic syndrome. More common in developed countries (usually seen as children, but also adults)
Etiology: Presents 1–6 weeks following infectious illness of streptococcus pharyngitis or impetigo (skin infection)
Pathogenesis: Circulating immune complexes (streptococcal ag and ab deposit within glomeruli and activate complement. Elevated antibodies against nephritogenic antigens in patients (AS0: antistreptolysin antibody). OR, Infection causes alterations of intrinsic proteins within the GBM –>ab to ag–> compelement
Diagnosis: Active urine sediment (dysmorphic RBCs, RBC casts); may have elevated antibody titers (as above); low C3 and total complement with normal C4 due to activation of alternative pathway. Streptozyme test measures 5 different antibodies.
Treatment: Care is supportive. Treat the underlying infection. Children have an excellent prognosis, with 95% recovering fully. Adults may have slow insidious progression to chronic glomerulonephritis.

Contrast the characteristic light (LM), immunofluorescence (IF) and electron microscopy (EM) features of:
IgA nephropathy
post-infectious glomerulonephritis.

IgA: LM: increased mesangium
IF: Mesangial IgA, C3
EM: Immune deposits mainly within mesangium
Post-infectious glomerulonephritis: LM: hypercellular glomeruli w/ abundant PMN’s
IF: granular deposits of IgG, C3
EM: Large, “hump-like” subepithelial deposits

Lupus nephritis
epidemiology
pathogenesis
course
treatment
Epidemiology: occurs in 50% patients w/ SLE (cause of morbidity/mortality); higher incidence in females and african americans (lower socioeconomic settings)
Pathogenesis: Autoimmune disease: ab’s directed at nucleosomes form immune complexes that deposit in kidney (form in circulation, from intrinsic renal ag, soluble nucleosomal proteins), because apoptosis cells not cleared from system properly.
Treatment: Class III-V are treated aggressively. Active lesions are treated with cyclophosphamide or cellcept + prednisone. Other immune therapies: Azathioprine (maintenance), Rituximab (anti-CD20 antibody) to target B-cells.
Describe the light, immunofluorescence and electron microscopy findings of the three most common patterns of lupus nephritis (Class III, IV and V).

LM: variable
Focal proliferative pattern (Class III): < 50% of the glomeruli in the sample show active lupus lesions (hypercellularity, immune deposits in capillary or mesangium ‘wire loops’
Class IV: >50% glomeruli show active lupus lesions
Class V: Membranous pattern. Has subepithelial immune deposits with spiking of the GBM (similar to membranous nephropathy)
“Full house” immune complex deposition: IgA, IgG (strongest), IgM, C3, C4, C1q (strong) in mesangium and capillary loops

What differentiates membranous lupus nephritis from idiopathic membranous nephropathy on renal biopsy
Membranous lupus nephritis has subepithelial immune deposits with deposits in the mesangium, without duplication of the GBM. There is “spiking” of the GBM. Foot processes are still present moreso than in membranous nephropathy (more hematuria than proteinuria).
Idiopathic membranous nephropathy has numerous small subepithelial immune deposits with effacement of the foot processes.
What is rapidly progressive glomerulonephritis (RPGN) and describe the three categories of disease leading to RPGN.
severe acute nephritic syndrome characterized morphologically by presence of necrosis and crescent formation on biopsy
Anti-GBM disease: acute nephritis, may have pulmonary-renal syndrome (Goodpasture’s syndrome)
Immune complex mediated disease: IgA nephropathy, Post-infectious GN, Lupus nephritis
Pauci-immune type: ANCA-associated glomerulonephritis, small vessel vasculitis that targets the glomerular capillaries
Anti-GBM
Etiology
Pathogenesis
Course
Treatment
Serologic tests

Etiology: Usually presents as Goodpasture’s in patients <30. In patients >50, just present with glomerulonephritis
Pathogenesis: formation of autoantibodies to type IV collagen
Course: Rapidly progressive. Needs immediate treatment (renal emergency)
Treatment: High dose steroids (IV methylprednisolone over 3 days), cyclophosphamide (3–6 months), azathiprine (maintenance), plasmapheresis
Serologic tests: Anti-GBM antibodies
Pauci-immune glomerulonephritis
Pathogenesis
Course
Treatment
Serologic tests
Pathogenesis: antineutrophil cytoplasmic antibodies (ANCA). No or few immune deposits. Pathogenesis poorly understood.
c-ANCA: target antigen - proteinase 3; associated w/ granulomatosis w/ polyangiitis
p-ANCA: target antigen - myeloperoxidase
Course: Rapidly progressive. Needs immediate treatment (renal emergency)
Treatment: High dose steroids (IV methylprednisolone over 3 days), cyclophosphamide (3–6 months), azathiprine (maintenance), plasmapheresis
Serologic tests: Anti-GBM antibodies, pANCA, cANCA
Compare/contrast the light, immunofluorescence and electron microscopy findings in anti-GBM disease and pauci-immune glomerulonephritis.

anti-GBM disease: LM: necrosis of glomerular capillaries and crescent formation. Non-involved glomeruli look normal.
IF: linear IgG along glomerular capillary walls
EM: no immune deposits present
pauci-immune glomerulonephritis: LM: there is necrosis of glomerular capillaries and crescent formation. Non-involved glomeruli look normal
IF: nothing lights up
EM: no immune deposits present
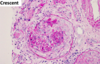
Understand the concept of chronic glomerulonephritis as evolution from multiple primary forms of glomerulonephritis, and describe the light microscopic appearance of advanced chronic injury (ESRD).

May be the result of many different etiologies that have progressed to ESRD/renal failure
LM: globally sclerosed, Interstitial fibrosis. atrophic tubules, arteriolosclerosis and hyalinosis.
Important point: Morphologic features of chronic GN are often not specific for underlying etiology. (impossible to determine primary cause because characteristic features are no longer present)

Acute Proliferative (Post Streptococcal) Glomerulonephritis
pathogenesis (use the name)
caused by immune complexes containing streptococcal antigens and specific antibodies, which are formed in situ.
arises after a B-hemolytic streptococcal infection of the skin (impedigo) or pharynx.
granular immune deposits in the glomeruli indicative of immune complex-mediated mechanism –> IgG, C3 deposits in mesangium and GBM.
enlarged/hypercellular glomeruli
“hump-like” deposits: deposit on subendothelial side –> GBM –> subepithelial side (*mechanism not understood)
total complement will be low/normal/high in the serum of Membranoproliferative Glomerulonephritis (MPGN)
LOW: C3, C4, and total complement
activates classical pathway
What regulates Ca2+ levels
PTH –> osteoblasts –> osteoclasts
PTH increases Ca reabsorption at DCT
Convert Vit D –> Calcitriol, indirectly increases Ca reabsorption in GI tract
Calcitriol effect on calcium
Calcitriol increases intestinal calcium absorption. The role in calcium excretion is unclear. (Calcitriol is the active form of Vitamin D, 1,25(OH)2D)
Regulators of Phosphate (3)
What inhibits NaPi2 in PCT
calcitriol does what to Phosphate?
Phosphate effect on FGF23
PTH –> reduced Phosphate reabsorption at PCT due to inhibition of Na+,phosphate cotransporter. PTH also increases phosphate release from bones. Net Effect = decreased serum phosphate
Calcitriol increases intestinal phosphate absorption, increases renal phosphate reabsorption
Phosphatonin (FGF 23). High serum phosphate increases FGF 23.
Pathogenesis of Secondary Hyperparathyroidism
Chronic Kidney Failure –> impaired Vit D formation –> impaired Phosphate excretion –> CalciumPhosphate binds and is excreted –> Hypocalcemia
Calcitriol stimualtes intestinal Ca2+ absorption –> low Calcitriol levels leads to Hypocalcemia –> stimulate PTH –> renal osteodystrophy –> Increased serum phosphate –> FGF-23 (inhibits calcitriol/ decrease phosphate reabsorption in PCT)
Vitamin D is converted by the proximal tubules of the kidney to the active form. As kidney function is impaired, active vitamin D formation is diminished. Phosphate excretion is impaired as well.
Calcitriol stimulates intestinal calcium absorption; low calcitriol levels lead to low levels of calcium absorption in the intestine and lead to low serum calcium levels.
Low serum calcium levels stimulate the parathyroid gland to release PTH. This response is mediated by the calcium sensing receptor.
Elevated PTH contributes to renal osteodystrophy
Increase in serum phosphorus causes an increase in FGF-23, which acts to decrease phosphate reabsorption in the proximal tubule. FGF-23 also inhibits calcitriol.
Clinical features of chronic kidney disease-mineral and bone disorder
causes
Abnormal circulating calcium/phosphorus leads to vascular calcification, as vascular smooth muscle cells are capable of producing bone-like proteins
Clinical features of kidney disease-mineral and bone disorders: bone pain, muscle weakness, skeletal deformities, growth retardation in children
Can be high turnover bone disease, in which PTH is high and the bone structure is disrupted; osteitis fibrosa has increased bone formation/resorption, increased osteoblast/osteoclast activation, peritrabecular fibrosis
Adynamic bone disease: low PTH, low bone turnover, decreased osteoblast/clast activity and low rate of bone formation, but mineralization is normal.
Osteomalacia: similar to adynamic, with mineralization defect
Mixed uremic bone disease: features of osteomalacia and osteitis fibrosa.

Uric Acid


Calcium Oxylate

Calcium Carbonate

WBC’s - abnormal in urine
seen in cystitis or pyelonephritis

Epithelial Cells - abnormal in urine
renal tubular or squamous cells

Dysmorphic RBC’s - abnormal urine
shearing of membranes, indicative of GBM in acute nephritic syndromes

Cystine

Tyrosine - abnormal in urine

Indinavir - Drug
Used for HIV and can precipitate –> leading to inflammatory response–> interstitial nephritis
Prevent: increase hydration, pH

Cholesterol - abnormal in urine

Struvite (Urea-splitting bacteria)
Precipitate in Alklaine urine forming Staghorn Calculus
When you see Casts in the Urine, where was this formed?
What do RBC casts mean?
PCT and TAL
Glomerulonephritis
You see WBC casts.
What usually causes this (3)
Tubulointerstitial disease (interstitial nephritis)
Pyelonephritis (infection)
Glomerulonephritis (w/ marked inflammation)
Granular (“muddy brown”) Casts are seen in?
Waxy Casts seen in?
Acute Tubular Necrosis (renal tubular cells)
Break-down of other cellular casts
Waxy (old): Chronic renal disease
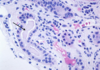
Hyaline Casts - nonspecific, seen in normal urine
They are seen in low numbers in normal people and are increased in glomerulonephritis, pyelonephritis, congestive heart failure, mental stress and strenuous exercise. They may also increase following use of certain therapeutic and chemical agents.

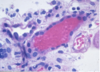
Red Blood Cell Cast
They are seen in diseases with damaged glomerular basement membranes and therefore indicate a highly significant glomerular injury. Diseases that cause acute glomerulonephritis, such as lupus nephritis and anti-GBM disease often have red blood cell casts, but they can also be seen with ischemic injury such as renal infarction


ATN: sequale of epithelial cells being sloughed off –> cells stretch so there is not ‘bare’ basement membrane
Epithelial Casts seen below

Muddy brown casts seen in ATN

Lipid and protein laden renal tubule seen in nephrotic syndromes
tubules trying to reabsorb as much protein as possible

WBC Casts seen in Pyelonephritis and Post-infectious glomulonephritis

Waxy casts are seen in Chronic Kidney Injury - urine stasis


Arteriosclerosis: intimal sclerosis (thickening) in response to DM and/or HTN

Arteriolar Hyalinosis (smaller vessels): many proteins get ‘stuck’ under the endothelium making lumen smaller. Occurs in DM (present in afferent/efferent arteriole) and HTN (more afferent arteriole)

Malignant Hypertension: endothelium becomes injured due to high BP –> intima swells and RBCs get trapped

Embolic Disease: athlerosclerotic plaques
Vasculitis: from implant rejection, will sometimes see Fibrinoid Necrosis! yiipee

Urinalysis findings in Acute Tubular Necrosis
Urine will contain: muddy brown casts, may have epithelial cell casts as well.
Specific Gravity may be high depending upon whether or not a chemical agent is the cause (contrast dye, IV meds)
Urinalysis findings in Urinary Tract Infections
Urine will contain RBCs (likely due to hemorrhage)
WBCs, bacteria, nitrites, and leukocyte esterase. May contain trace protein
Urinalysis findings in Allergic Interstitial Nephritis
Urine will contain eosinophils and WBC casts (eosinophils, likely)
Urinalysis findings:
Nephrotic syndrome vs. Acute Nephritic Syndrome
Nephrotic: proteinuria > 3.5g/24hr, with NO hematuria or glucose. Fat oval bodies may be present
Nephritic: proteinuria may be present but usually < 3.5g/24hr, with hematuria, RBC casts, dysmorphic RBCs (characteristic of glomerular injury)
How does Gout lead to Uric Acid stone formation
what would the urinalysis look like?
What drug would decrease uric acid formation
Uric Acid crystals would be found in the urine, as well as RBCs and WBCs.
Allopurinol, febuxostat (inhibitor of xanthine oxidase)

Why are Struvite Stones clinically significant

occur as a result of a bacterial infection. “Coffin lid” crystals.
Struvite (presumably because magnesium and sulfur - these are dense atoms that will not allow x-rays to penetrate through; unique in that they occur at an alkaline pH)
Why are Indinivar stones clinically significant?
What does this drug do?

Indinavir crystals: needle shaped crystals with irregular borders, often arranged in a fan shape. They precipitate out of the urine in people taking the drug Indinavir –> nephrolithiasis. They damage the tubular cells and provoke an inflammatory response.
-navir: Protease inhibitor: Inhibit maturation of new virus by blocking protease in progeny virions
What are the most common types of organisms associated w/ formation of struvite stones
How does urine pH influence stone formation?
- Proteus mirabilis*
- Klebsiella*
- Ureaplasma*
Urease hydrolyzes urea to release ammonia and CO2 –> High pH (alkaline). Predisposes to struvite (ammonium magnesium phosphate) stones, particularly
How to calculate Respiratory compensation in Metabolic Acidosis
pCO2 = (1.5 X HCO3) + 8 +/-2
or pCO2 = last 2 digits of pH
How to calculate the Respiratory compensation in Metabolic Alkalosis
pCO2 = variable increase or
pCO2 = (0.7 X HCO3) + 20
How to calculate renal compensation in Respiratory Acidosis
acute and chronic
Acute: HCO3 increases 1 mEq/L for every 10 mmHg increase pCO2
Chronic: HCO3 increases 3.5 mEq/L for every 10mmHg increase pCO2
How to calculate renal compensation in Respiratory Alkalosis
acute and chronic
Acute: HCO3 decreases 2 mEq/L for every 10 mmHg decrease pCO2
Chronic: HCO3 decreases 5 mEq/L for every 10 mmHg decrease pCO2
Causes of Metabolic Acidosis w/ Elevated Anion Gap
“MUDPILES” and “MUDPALES”
Methanol
Uremia
DKA
Propofol
Iron/ Alcoholic Ketoacidosis
Lactic Acidosis
Ethylene Glycol
Salicylates
Metabolic Acidosis w/ Normal ANION gap
Causes:
Renal tubular acidosis
Diarrhea
Carbonic Anhydrase Inhibitors
Administration of HCL or NH4+
Overview of of PKD pathophysiology
Treatment
- Role of primary cilia in PKD
– intracellular Ca and cAMP
– increased proliferation/apoptosis
- For ADPKD, cystogenesis is a two-step process – cyst initiation: earlier in PKD1 than in PKD2 – cyst expansion: faster in males than females.
- Critical issues in managing PKD*:
– Screening relatives at risk
– Blood pressure control
- Potential treatment:
– V2R antagonists reduce cAMP levels and slow cyst growth in animals with PKD
