Introduction to CNS pharmacology Flashcards
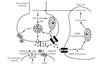
Diagram of Chemical Neurotransmission

Diagram of when neurons reach a threshold potential.
What’s notable about the discovery of drugs which treat CNS disorders?
Much of the discovery of drugs to treat CNS disorders has been serendipitous (i.e. based on luck)
Give a few examples of serendipity in CNS drug discovery.
- The first antiepileptic drug (phenobarbital) was prescribed as a sedative but had anticonvulsant properties
- The first antipsychotic drug (Thorazine) was also prescribed as a sedative but stopped hallucinations
- The first antidepressant drug (imipramine) came from a failed attempt to develop a new antipsychotic drug
What are astrocytes?
Astrocytes maintain nutrition and regulate ionic concentrations. They also play a role in neurotransmitter synthesis and metabolism.
What do Oligodendrocytes do?
Oligodendrocytes (& Schwann cells in the periphery) produce myelin (inset) that insulates nerve cell membranes.
What is the function of Microglia?
Microglia act like macrophages, scavenging unwanted materials from the brain. They proliferate in disease states.
What are some of the neurotransmitters in the CNS?
Amines
- Dopamine
- Serotonin
- Noradrenaline
Amino Acids
- Dopamine
- Serotonin
- Noradrenaline
- Histamine
Cholinergic
- Acetylcholine
Purines
- ATP
- Adenosine
Peptides
- Enkephalins & endorphins
- Vasoactive intestinal polypeptide (VIP)
- Substance P
- Cholecystokinin (CCK)
- Opioids
Some neurotransmitters have well-defined roles (e.g. glycine), but many have complex and overlapping functions (e.g. amines in emotion and cognition).
How are neurons defined?
Neurons are largely defined by the neurotransmitter that they release.
List of receptors and the receptors subtypes.
Dopamine: D1, D2, D3, D4, D5
Serotonin: 5HT1A/1B/1D/1e/1F, 5HT2A/2C, 5HT3A/3B/3C/3D/3E, 5HT4, 5HT5A, 5HT6, 5HT7
Noradrenaline: a1A/1B/1D, a2A/2C,b1, b2
Glutamate: AMPA (GluA1-4), NMDA (GluN1, GluN2A-D, GluN3A/3B), kainate (GluK1-5), metabotropic (mGlu1-8)
GABA: GABAA (a1-6, b1-3, g1-3, d, e, q, p, r1-3), GABAB1/B2
Acetylcholine: nicotinic (a2/3/4/5/6/7/9, b2/3/4), muscarinic (mostly M1, M4, M5)
Purines: A1, A2A, A3, P2X1/2/3/7, P2Y1/2/4/6/11/12/13/14
Tachykinins: NK1, NK2, NK3
Opioids: u, k, d, ORL1
Impossible to remember them all but try to remember those that are ligand-gated ion channels and those that are GPCRs.
What are some of the technical issues with neuropharmacology?
Technical issues
- Tissue baths cannot be used, so instead researchers use:
- Electrophysiology
- Lesion studies
- Genetic techniques (KO mice, siRNA, etc.)
- Behavioral effects of selective drugs
- Very few reliable animal models; can’t biopsy tissue
- Outputs are hard to measure both in humans and animal models (although for different reasons)
- Using induced pluripotent stem cells derived neurons to model brain diseases
Most of the techniques above are NOT unique to neuropharmacology (e.g. electrophysiology); the difference mainly lies in what cannot be done in the brain.
Describe the plasticity within the CNS?
- The brain (and its pharmacology) continues to develop into adulthood
- The information the brain stores and processes change constantly
- Continued post-natal migration of cells and development of connections
- Changes in the strength of connections throughout life, e.g. long-term potentiation & depression
- Neurogenesis (in specific brain regions)
What is the Blood-Brain Barrier?
The blood-brain barrier (BBB) is a crucial immunological feature of the human central nervous system (CNS). Composed of many cell types, the BBB is both a structural and functional roadblock to microorganisms, such as bacteria, fungi, viruses, or parasites, that may be circulating in the bloodstream.
Give some of the features of the Blood-Brain Barrier.
- Continuous endothelium
- Tight junctions & basal membrane
- Lack of fenestrations
- Pericytes and astrocytes
- Few pinocytotic vesicles
- High metabolic rate
- Drug transporters
- Drug metabolizing enzymes
- The brain is inaccessible to many drugs (~85%); the drug requires high lipid solubility.
- The BBB is weak at the postrema
- During a brain infection, the Blood-Brain Barrier is compromised.
Types of Dementia
- Alzheimer’s disease (>50% of cases)
- Multiple cerebral infarcts (Vascular dementia) 20%
- Dementia with Lewy bodies (15%)
- Frontotemporal dementia (5%)
What are some causes of dementia?
- Parkinson’s/Huntingdon’s/Pick’s disease
- Prion disease (e.g. CJD)
- Alcoholism (Wernicke-Korsakoff), syphilis, HIV
- Other damage/metabolic or vit B deficiency etc
What’s the most common form of senile dementia in the USA population incidence?
0.1% at 60 (early onset)
Rising to 10% by age 80 (Late-onset)
Rising to over 30% by age 90 (Alzheimer’s Disease)
Alzheimer’s symptoms.
- Onset may be very slow and gradual or rapid
- 4-12 years(7-10 most common)
‘Gradual onset and continuing decline of cognitive function from a previously higher level resulting in impairment of social and occupational function’
- Decrease in the following:
- Memory
- language
- Perceptual abilities
- 1st symptom = loss of recent memory and inability to store recent information in long term memory
Common Symptoms of Alzheimer’s by stage.

The Aetiology of Alzheimer’s
A number of environmental and genetic risk factors may be involved e.g.
- head trauma
- exposure to aluminium
- family history of Down’s syndrome
- cerebrovascular disease
- Increasing age
- Gender-specific? ( 2x at risk thanbut live longer!)
- Genetic mutation/s
Describe the Neuropathology when the brain is in the disease state of Alzheimer’s.
- Neurone degeneration and death occurs in the hippocampus & the amygdala (subcortical limbic system)
- mainly regions of the brain that function in memory and learning.
- Massive cell loss and brain shrinkage
- Atrophy in parts of the cerebral cortex
- Thinned temporal lobes and ventricle size
What is the Limbic System?
The limbic system is the part of the brain involved in our behavioural and emotional responses, especially when it comes to behaviours we need for survival: feeding, reproduction and caring for our young, and fight or flight responses.
What does the limbic system consist of and how is it affected by Alzheimer’s?
The limbic system consists of a number of structures, including the fornix, hippocampus, cingulate gyrus, amygdala, parahippocampal gyrus, and parts of the thalamus. The hippocampus is one of the first areas affected by Alzheimer’s disease. As the disease progresses, the damage extends throughout the lobes.
Diagram of the Gross Anatomical Pathology of Alzheimer’s

What is the pathology of early and late-onset Alzheimer’s?
- Senile or amyloid plaques in the brain
- deficits in amyloid processing
- Presence of neurofibrillary tangles in the cerebral cortex
- Amyloid microangiopathy
- Glial inflammation and toxicity
- Leading to the Amyloid Cascade Hypothesis
What is the amyloid cascade hypothesis?
The amyloid cascade hypothesis postulates that the neurodegeneration in AD is caused by the abnormal accumulation of amyloid-beta (Aβ) plaques in various areas of the brain.
“deposition of amyloid β protein (AβP), the main component of the plaques, is the causative agent of Alzheimer’s pathology and that the neurofibrillary tangles, cell loss, vascular damage, and dementia follow as a direct result of this deposition”
Research into various approaches and therapies has centered around this cascade theory – unfortunately with little success to date.
Stages in the Amyloid Cascade Hypothesis.

What are Amyloid Plaques?
- The amyloid plaques contain an insoluble 42-residue peptide called Ab42 (beta-amyloid 42) in core
- surrounded by axons/dendrites (neurites) & microglia & astrocytes
- structural abnormalities include enlarged mitochondria, liposomes & impaired filaments
- It May take decades for plaques to ‘mature’
- Contain glial cells- microglia, astrocytes
- N.B these plaques are not specific for AD but more prevalent

How is beta-amyloid 42 derived?
- 42 amino acid’s long
- (In a healthy brain 90% of the Ab peptide is in the Ab40 form)
- Derived by cleavage of the much larger protein encoded by chromosome 21
- Amyloid precursor protein (beta-APP)
- Normally cleaved by enzyme (secretase)à Ab40 form
How are Beta-Amyloid Plaques formed?
The amyloid precursor protein (APP) is a transmembrane protein that can undergo a series of proteolytic cleavages by secretase enzymes. When it is cleaved by α-secretase in the middle of the β-amyloid domain (Aβ), it is not amyloidogenic. However, when APP is cleaved by β-and γ-secretase enzymes, neurotoxic Aβ peptides are released, which can accumulate into oligomer aggregate.
Mutations in the APP gene tend to inhibit cleavage by α-secretase and consequently enable preferential cleavage by β-secretase. Mutations in the presenilin-1 and presenilin-2 genes (PSEN1 and PSEN2), which are components of the γ-secretase complex, increase cleavage by γ-secretase at this site. In both situations, the result is excess Aβ peptide production.

What does the current Beta Amyloid Hypothesis suggest?
The current Aβ hypothesis suggests that the soluble oligomers can impair synaptic function between neurons. Simultaneously, the oligomers may aggregate into insoluble β-sheet amyloid fibrils, which can trigger a local inflammatory response. 22 Over time, the subsequent oxidative stress and biochemical changes ultimately lead to neuronal death and the development of neuritic plaques typical of Alzheimer’s disease.
Aβ42 peptide – evidence for………
- detrimental memory formation
- initiation of Tau phosphorylation
- associated with demyelination
- tangle formation
- oxidative damage
- inflammatory responses (microglia)
- deficits in NT’s (excess glutamate)
- apoptotic cell death
What are Neurofibrillary Tangles?
- Dense bundles of fibres in cytoplasm of neurones
- Neurofibrillary tangles contain a highly polymerised form of a cytoskeletal protein called tau in cytoplasm
- Occur in many chronic brain diseases
- Many people who live to late 70’s will have both senile plaques and neurofibrillary tangles
- Hippocampus & parieto-temporal regions of cerebral cortex particularly susceptible.
Microtubules are tracks that transport nutrition and other molecules. Tau-proteins act as “ties” that stabilize the structure of the microtubules. In AD, tau proteins become hyperphosphorylated and tangled, destabilizing the structure of the microtubule. Loss of axonal transport results in cell death.
Stages of Hyperphosphorylation of tau.
- Hyperphosphorylation of tau
- Tau destabilizes and detaches from microtubules
- Paired helical filaments form neurofibrillary tangles
- Microtubules destabilize leading to disintegration

Apolipoprotein E and its link to Alzheimer’s.
- Apolipoprotein E -neuronal repair and growth (cholesterol transport)
- Common amino acid variations are also shown to be associated with a predisposition to Alzheimer’s disease
- However, also detected in both senile plaques and neurofibrillary tangles
- in vitro, it has an affinity to Ab42 peptide-
- increases formation
- interferes with removal
What are the 3 major Alipoprotein E alleles?
Three major APOE alleles
- APO*E2 (approx 6% of population)
- Protection from Alzheimers
- APO*E3 (approx 78% of population)
- APO*E4 (approx 16% of population)
- increases risk of Alzheimer’s in a dose dependent fashion.
- the alleles differ by just two amino acids
How does APOE4 result in an increased risk of developing AD?
- Apolipoprotein E gene on Ch 19 is required for normal cholesterol transport necessary for removal of β amyloid protein from CNS
- So APOE4à accumulation of β amyloid + binding of APOE protein to τ protein of neurofibrillary tangles
- Heterozygous APOE4à 2 x risk of AD
- Homozygous APOE4à 5 x risk of AD
Statistics about Familial ALzheimer’s
- 5% of families clearly show autosomal dominant form of inheritance
- Most of these families early onset
- However, only 30% of early-onset cases show autosomal dominant inheritance
- 34% of overall risk of Alzheimer’s is probably due to hereditary factors
What are some of the neurotransmitter systems affected?
- Acetylcholine
- Synthesised by the basal forebrain cholinergic complex (BFCC) axons project to the hippocampus & the cerebral cortex
- 40-90% ¯ChAT (acetylcholine transferase)
- Glutamate
- b amyloid protein accumulates in neuronesà increased release of glutamate
- High levels disrupt learning and memory (NMDA receptors)
- Excitotoxicity
- Catecholamines, somatostatin, corticotrophin etc
What are some of the treatments available for AD?
- Target Ach & glutamate (memory) & help disability
- Block Ach breakdown by inhibiting Acetylcholine esterase (AChE)
- e.g. tacrine (Cognex),Doneprezil,Aricept
- Memantine (NMDAR antagonist N-methyl-d-aspartate) shown to reduce clinical symptoms with moderate to severe AD
Inconsistent & treating symptoms rather than cause
What is the Synaptic Plasticity and Memory Hypothesis?

What is a contemporary approach to the treatment of AD?
- Reduce activity of b amyloid
- b-site APP cleaving enzyme (BACE) inhibitors (block b secretase) & g-secretase inhibitors
- Problem: g-secretase is not specific for amyloid cleavage
- Disrupts lymphocyte development
- Affects intestinal structures
- BACE inhibition is not associated with toxic side effects but can’t cross BBB!
Inhibitors of amyloid aggregation.
- Inhibition of Aβ production and aggregation, acceleration of Aβ clearance as well as reduction of tau hyperphosphorylation.
- GAGS sulfated glycosaminoglycans bind A b in solutionà plaques
- GAG mimetics block aggregation process
- Ovine colostrinin (O-CLN)-improves learning in AD in animal models
SALAs and Alzheimer’s
- Selective amyloid lowering agents
- New class of anti-Ab drugs targeted at mild AD
- e.g. tarenflurbil modifies secretase which lowers Ab42
Why did tarenflurbil fail?
What are the reasons for this setback after the previous apparently encouraging results in a Phase II study? A straightforward explanation of this failure is that the γ-secretase is not the right target for therapy or that, in general, blocking Aβ does not produce clinical benefits in AD. If one still accepts the physiopathological role of Aβ in AD, tarenflurbil could not be the right compound because of its weak pharmacological activity as an Aβ1-42 lowering agent and its poor brain penetration.
What is Parkinson’s Disease?
A progressive disease of the nervous system marked by tremor, muscular rigidity, and slow, imprecise movement, chiefly affecting middle-aged and elderly people. It is associated with degeneration of the basal ganglia of the brain and a deficiency of the neurotransmitter dopamine.
What is the Basal Ganglia?
The “basal ganglia” refers to a group of subcortical nuclei responsible primarily for motor control, as well as other roles such as motor learning, executive functions and behaviors, and emotions. … Disruption of the basal ganglia network forms the basis for several movement disorders.
What are the three types of nuclei that the Basal Ganglia consist of?
- Input Nuclei
- Output Nuclei
- Intrinsic Nuclei.
What does the input nuclei in the Basal ganglia consist of?
The Input Nuclei consist of the striatum, which consists of the caudate and the putamen.

What does the Output Nuclei in the basal ganglia consist of?
Output Nuclei
- Globus Pallidus Internal
- Substantia Nigra
- Pars Reticulata

What does the Intrinsic Nuclei in the basal ganglia consist of?
Intrinsic Nuclei
- Subthalamic Nucleus
- Globus Pallidus external
- Substantia Nigra Pars Compacta

Basal Ganglia Circuitry Diagram

What does the Basal Ganglia do?
- Damage to any basal ganglia structure may cause slowness of voluntary movement, involuntary movements, involuntary postures, or a combination.
- Damage to the striatum causes voluntary movements to be slow and may produce involuntary movements.
- Damage to the STN causes large-amplitude involuntary movements.
- Damage to the GP causes slowness of movement, abnormal postures, difficulty relaxing muscles.
- Damage to SNpc causes tremor at rest, slowness of movement, rigidity and postural instability.
Basal Ganglia Motor Loop
How many people over the age of 65 does Parkinson’s Disease affect?
- Parkinson’s disease affects 1% of people over 65.
- Second most common neurodegenerative disease after Alzheimer’s disease.
Which sex has an increased prevalence of PD?
Increased prevalence in men.
What factors contribute to the onset of PD?
Environmental and genetic factors contribute to onset. In most cases idiopathic.
What are the characteristic symptoms of PD?
- Characteristic symptoms/signs occurring unilaterally then bilaterally:
- Tremor – occurs in 75% of PD patients
- Bradykinesia
- Muscular rigidity
- Impairment of postural reflexes
Diagram of the stages of Parkinson Disease

What causes the motor impairments in PD?
- Motor impairments are a result of the depletion of dopaminergic neurons within the substantia nigra pars compacta (SNpc).
- 60% of nigral neurons are lost before motor impairments appear.
- Delay accounted for by:
- a) increased dopamine production by surviving neurons
- b) upregulation of dopamine receptors in target striatal neurons.
- Nigrostriatal tract degenerates leading to less than 20% dopamine levels in basal ganglia.
What are some of the Dopamine Pathways?

Describe the Nigrostriatal Pathway
Motor control
Cell bodies within the substantia nigra, axons project to the striatum.
Accounts for 75% of dopamine in the brain.
Describe the Mesolimbic pathway.
- Emotional behaviour
- Cell bodies within the midbrain ventral tegmental area (adjacent to substantia nigra), project to the nucleus accumbens and amygdala.
Describe the Mesocortical pathway
- Emotional behavior
- Cell bodies within the ventral tegmental area, axons project to the frontal cortex.
Describe the Tuberohypophyseal pathway
- Endocrine control
- A group of neurones projecting from the ventral hypothalamus to the pituitary to regulate pituitary secretions.
What are the 2 different types of Dopamine Receptors?
There’s the D1 type and the D2 type.
The D1 type consists of the D1 and D5 receptors. The D2 type consists of the D2, D3, and D4 receptors.
Table of the distribution and functional roles of the D1 and D2 type receptors.
- Five different dopamine receptors were identified.
- All belong to the family of G-protein coupled transmembrane.
- D receptors are expressed in distinct but overlapping brain areas.
- Dopamine acts pre- and post-synaptically.
- D receptors also mediate effects peripherally: renal vasodilation and increased myocardial contractility.

What’s the pathology of Parkinson’s Disease?
- In 1912 Friedrich Lewy discovered ‘spherical neuronal inclusions’ during postmortem.
- Lewy bodies are defining feature of PD.
- Lewy bodies are neuronal intracytoplasmic inclusions comprising numerous proteins, including α–synuclein.
- Lewy bodies found in cytoplasm of surviving neurones.
What is the main constituent of Lewy Bodies? And what can the main constituent’s role be?
The main constituent of Lewy Bodies is α–synuclein. α–synuclein is a synaptic protein present in presynaptic terminals. Possible involvement in neurotransmitter storage and release, vesicle recycling, and synaptic plasticity. Mutations in synuclein gene identified in familial forms of PD.
What’s the proposed explanation for the loss of smell and the REM sleep disorders before the loss of motor function?
- Proposed that Lewy bodies deposited first in the olfactory bulb and lower brainstem, then in SN, progressing to the cortex.
- Explains loss of smell and REM sleep disorder onset before motor impairments.
What other dysfunction relating to PD has been identified in sporadic PD?
Mitochondrial dysfunction
- Mitochondrial complex 1 defect in brain confined to substantia nigra.
- Identified in sporadic PD
- Linked with oxidative stress and elevated brain iron levels.
What is post-mortem evidence of inflammation in PD?
Microgliosis
- Postmortem evidence of inflammation
- Change in cytokine levels in substantia nigra and CSF.
- Glial cell activation but is this triggering cell death or a response to cell death?
What are some of the causes of PD?
- Age: possible increased vulnerability of dopaminergic neurones to toxic insult because of age-related failure of physiological and biochemical processes.
- Several gene mutations have been identified in familial and sporadic PD.
- Environmental risk factors likely contribute but no specific agent has been identified as causative.
- Industrialisation, pesticides, rural environment, bacterial, viral?

Flowchart for the treatment of PD

Describe how Levodopa treats PD
- The first line of treatment.
- Dopamine does not cross BBB so precursor Levodopa is given.
- Levodopa converted to dopamine in dopaminergic neurones.
- Plasma half life is short – 2 hours.
- Given with a peripherally acting dopa decarboxylase inhibitor (e.g. carbidopa) to reduce peripheral side effects.
- Decarboxylase inhibitors do not cross BBB so decarboxylation occurs rapidly in the brain.
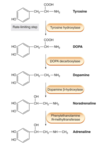
What are some of the pitfalls of Levodopa as a PD treatment?
Levodopa effectiveness decreases as PD advances.
Reducing ‘off time’ and dyskinesias (involuntary movements).
Require more continuous dopaminergic stimulation to a more constant physiological level.
Describe how Dopamine agonists treat PD?
Dopamine agonists
- Effective in controlling symptoms of PD, usefulness is limited by side effects: nausea, vomiting, somnolence, fibrotic reactions in lungs and pericardium.
- Do not show fluctuations in efficacy associated with levodopa.
- May cause somnolence, hallucinations and predisposition to compulsive behaviour e.g., excessive gambling.
Describe how MAO-B inhibitors treat PD.
MAO-B inhibitors
- Inhibit breakdown of extraneuronal dopamine in the brain.
- Lack unwanted side effects of non-selective MAO inhibitors used to treat depression.
- Clinical trials show the combination of MAO-B inhibitor Selegiline and levodopa is more effective than levodopa alone.
Describe how Neural transplantation treats PD.
Neural transplantation
- PD first neurodegenerative disease for attempted neural transplantation.
- Injection of foetal neuroblasts directly into the striatum.
- Some transplants relatively successful – functional dopaminergic connections.
- Development of serious dyskinesias.
- Five or more foetuses are required for one transplant.
Describe how Gene therapy treats PD.
Gene therapy
- Aimed at increasing the synthesis of neurotransmitters and neurotrophic factors.
- Dopamine in the striatum by expressing tyrosine hydroxylase or dopa decarboxylase.
- GABA in the subthalamic nucleus by overexpression of glutamic acid decarboxylase (to reduce excitatory input to SN).
How does deep brain stimulation treat the symptoms of PD?
Deep brain stimulation
- Treats PD symptoms.
- Not a cure
- Side effect: depression
- Not for everyone.
- Used in patients with motor complications – frequent ‘off periods’ while taking levodopa.
- Electrodes implanted during awake surgery into the subthalamic nucleus or globus pallidus.
What is epilepsy?
An episodic disorder of the nervous system arising from the excessively synchronous and sustained discharge of a group of neurons.
These discharges produce “seizures” that vary from one person to another in frequency and form.
What may a seizure manifest as?
A seizure may manifest as:
- a brief stare
- a change of awareness
- a convulsion
A seizure may last a few seconds or a few minutes.
What is the definition of a seizure?
The clinical manifestation of an abnormal and excessive excitation of a population of cortical neurons.
What is the definition of an ‘Epileptic Seizure’?
A broad category of symptom complexes arising from … recurrent paroxysmal episodes of brain dysfunction manifested by stereotyped alterations in behavior.
What is the definition of Epilepsy/
disorder of the brain characterized by enduring predisposition to generate epileptic seizures and by the neurobiological, cognitive, psychological, and social consequences of this condition
More than 50 different epilepsies are recognized
What are the classifications of epilepsies by seizure type?
There are:
- Partial Seizures
- Generalized Seizures
- Unclassified Epileptic Seizures
What seizures fall under the Partial (focal) seizures classification?
- Simple Partial Seizures - awareness is not impaired
- Complex Partial Seizures - awareness is impaired
- Partial Seizures evolving to Secondarily Generalized Seizures.
What seizures fall under the Generalized Seizures Classification?
- Absence Seizures
- Myoclonic Seizures
- Clonic Seizures
- Tonic Seizures
- Tonic-Clonic Seizures
- Atonic Seizures
Diagram of the brain in the epilepsy disease states. From Partial Seizures - Unclassified epileptic seizures.

Pie Chart Demonstrating the Prevalence of Seizure types.

How many people does epilepsy affect world-wide and in the UK?
Epilepsy affects more than 50 million people worldwide, and more than 500,000 people in UK.
What is the % lifetime risk of a single unprovoked seizure?
The lifetime risk of a single unprovoked seizure is 2% - 5%.
What is the prevalence of active epilepsy?
The prevalence of active epilepsy is 0.4% - 1.0%.
The prevalence of active epilepsy increases with age.
What percentage of epilepsies are presumed to have a genetic cause?
- Around 40% of all epilepsies are presumed to be genetic*
- minority (2-5%) show monogenic inheritance (single gene defect)
- the majority (>90%) assumed to have polygenic inheritance (multiple gene defects)
- an increasing number (2-5%) have de novo mutations (not inherited)
What percentage of epilepsies are associated with an antecedent brain injury?
- Another 30% of epilepsy cases are associated with an antecedent brain injury
- birth trauma (e.g. peri-natal hypoxia)
- aftermath of infection (e.g. meningitis)
- head trauma (e.g. car accident, sports injury, etc.)
- cerebrovascular malformation
- brain tumour
- stroke
What percentage of epilepsy cases are idiopathic?
30% of epilepsy cases are idiopathic.
What happens in the brain during epileptic episodes?
The simple answer is that we don’t know.
It probably involves:
- Ion channel dysfunction
- Neurotransmitter imbalance
- Other things as yet unknown
They may be caused by:
- Genetic Mutations
- Damage to the brain
- Other things as yet unknown
Diagram of Ion Channels in Epileptic Discharges

How are the neurons arranged in the brain?
Neurons are arranged in complex networks; 1011 cells forming 1015 synapses.
What does cell-cell signaling require?
Cell to cell signaling requires the release of neurotransmitters from nerve terminals
What do NTs act?
NTs act on receptors in pre- and post-synaptic membranes.
What contributes to excitability in neuronal networks?
Neurotransmitter receptors contribute to excitability in neuronal networks.
What are the two main NTs in epilepsy? And describe what they act on and if they generate EPSPs or IPSPs.
The two main NTs in epilepsy are:
Glutamate and GABA.
Glutamate
- Glutamate is the principal excitatory NT in the mammalian brain.
- It acts on AMPA, KA, and NMDA receptors, leading to Na+ and Ca2+ influx.
- Generates excitatory post-synaptic potentials (EPSPs)
GABA
- GABA is the principal inhibitory NT in the mammalian brain
- Acts on GABAA (and GABAB) receptors, leads to Cl- influx
- Generates inhibitory post-synaptic potentials (IPSPs).
What are some of the other neurotransmitters which are less well characterised in epilepsy?
Dopamine, Serotonin and Acetylcholine.
If epilepsy is caused by a neurotransmitter imbalance between Glutamate and GABA, how would it look?
In the normal brain, there is an equal balance between Glutamate and GABA.
It is possible that epilepsy can be caused by an excess of Glutamate compared to the concentration of GABA.

What is SMEI? What are the symptoms?
- SMEI is a form of monogenic epilepsy, which stands for Severe Myoclonic Epilepsy of Infancy (aka Dravet syndrome).
- Children present at ~6 months with generalised clonic seizures;myoclonic, absence and focal seizures may develop later (1-4 years).
- The seizures are typically unresponsive to antiepileptic drugs; children develop cognitive, behavioral, and motor impairments.
What is the mutation involved in SMEI?
Mutations in SCN1A gene - the Nav1.1 subunit of the voltage-gated sodium channel.
- 100s of mutations (often de novo) are now described in the SCN1A gene.
- 70-80% of SMEI cases carry an SCN1A mutation
These mutations eventually lead to the loss of function (unlike GEFS+).
How could the loss of the SCN1A gene possibly lead to SMEI?
- Loss of NaV1.1 leads to compensatory up-regulation of other sodium channels?
- NaV1.1 predominantly expressed in inhibitory interneurons; loss of inhibitory control?
- Sodium channel blocking drugs make SMEI worse; block residual channel function?
How did Tracy Dixon-Salazar treat her daughter’s De novo mutation-based epilepsy
Savannah Dixon-Salazar
- No family history of epilepsy
- Developed Lennox-Gastaut syndrome at 2 years
- All drugs ineffective - 300 seizures per month
- Mum (Tracy) did BSc in Neuroscience, PhD in Genetics
- Exome-sequenced Savannah’s DNA
- Identified 25 novel calcium ion channel variants
- Treated with Ca2+ blocker verapamil (anti-arrhythmic drug)
- Seizures down to 20/month; now only at night
Intellectual development hugely improved
What are symptomatic/acquired epilepsies usually? And what may they be associated with?
Acquired or symptomatic epilepsies are often focal seizure disorders. Discernable abnormality is often ID’d on MRI scan. May be associated with previous:
- perinatal hypoxia
- complex febrile seizures
- brain infection
- status epilepticus

Diagram of Networks and Synaptic Connections
What are some theories for hyperexcitable networks in epilepsy?

What are some of the consequences of epilepsy?
- Remains subject to considerable stigma
- Uncontrolled seizures in one-third of patients
- Cognitive decline in some patients, depression, anxiety
- Loss of driving license, loss of job, loss of social life?
- In general - lower educational attainment, less likely to marry
- Dependent behavior – impact on families and carers
- Risk of injury
- Mortality: 3x higher than the general population, 9x in uncontrolled epilepsy
Who synthesized Phenobarbital?
Synthesized as a sedative by Emil Fischer in 1911. It was marketed by F. Bayer & Co. as “Luminal”.

What was an interesting effect of Phenobarbital?
When administered to patients with epilepsy, they had become seizure-free.
When was Valproic acid synthesized?
First synthesized in 1882 by Burton but there was no known use. It is liquid at room temperature so was used as a lipophilic vehicle for water-insoluble compounds.
How did Valproic acid show efficacy against PTZ seizures?
- Pierre Eymard (Grenoble, France) used VPA to dissolve khelline derivatives for Ph.D. studies in 1962
- Vehicle (i.e. VPA) but not khelline derivatives showed efficacy against PTZ seizures in mice & rabbits
- Earliest clinical trials in 1964, marketed for the treatment of epilepsy in France in 1967
Describe Voltage-Gated Sodium Channels and what they’re responsible for?
- Apart of the super-family of voltage-gated channels; form ion-selective pores in the membrane
- Responsible for depolarisation of the nerve cell membrane and conduction of action potentials across the surface of neuronal cells
- Expressed throughout the neuronal membrane, on dendrites, soma, axons, and nerve terminals; expression is highest in the axon initial segment (AIS)

What are the drugs Phenytoin and carbamazepine examples of? And how do they function?
Phenytoin and carbamazepine are classical sodium channel blockers. They show preferential binding to inactivated state of the channel.
They exert a ‘use-and frequency-dependent block of channel function.
The block is enabled by the use (i.e. activation) and enhanced by the frequency of use. So high-frequency action potential firing is preferentially reduced (i.e. during a seizure).
Low-frequency firing and single action potentials are largely unaffected.
Other Anti-epileptic Drugs have similar effects - subtle differences only.
What is the principal inhibitory NT in the mammalian brain?
And what % of synapses is it released at?
Gamma-aminobutyric acid (GABA) is the principal inhibitory neurotransmitter in the mammalian brain. It is released at 40% of all synapses.
What are the two GABA receptors and which are responsible for fast inhibition and for slower inhibition?
- GABAA receptor – fast inhibition
- GABAB receptor – slower inhibition
How is the reuptake of GABA facilitated?
The reuptake of GABA is facilitated by GAT (GABA transporter) on the Glial cell back into the pre-synaptic cell as Glutamine and converted into GABA again.
How is GABA converted into Glutamine again?
GABA transaminase converts it into Glutamate which then gets converted into Glutamine.
Diagram of GABAergic neurotransmission