hematology Flashcards
lifespan of blood cells
o Erythrocytes=120d
o Granulocytes=0.5d
o Platelets=9d
sites of hematopoeisis throughout the lifespan
o Prenatal: yolk sac
o 4months gestation—birth: liver, spleen
o Childhood: bone marrow (all over, incl tibia/femur)
o Adult: bone marrow shifts to vertebrae, sternum, and ribs predominantly
o *w/ inc. demand for blood cells, hematopoeisis can persist or be reestablished in normally inactive marrow sites (even liver/spleen)
bone marrow architecture
o Cords (niches)
• Mixture of cell types in various stages of maturation
o Sinuses
o Bone marrow-blood barrier
o Adipose and fibroblast cells and their secretions (adhesive molecules and chemokines, like CXCL12) provide the special microenvironment that allow HSC to survive (homing)
3 requirements for hematopoiesis
o Stem cells
o Stroma/extra-cellular matrix (microenvironment)
o Growth factors (regulators)
growth factors in hematopoiesis
o Stem cells (stem cell factor factors)
o Myeloid cells (GM-CSF, G-CSF, M-CSF)
o Erythroid cells (erythropoietin)—epo synthesized by kidney cells in response to hypoxia
o Megakaryocytes (thrombopoietin)
o Lymphoid cells (interleukins, cytokines)
o Growth factors (except EPO) exhibit redundancy (overlappin functions), pleiotrophy (multiple functions; stimulate multiple cells), and synergy (combo are more effective than individual factors)
o Synthesis is high localized w/ GF tethering
o Myeloid GF influence primitive progenitor cells and mature progeny
o Act to: maintain cell viability, initiate cell cycle, activate effector functions
hematopoietic stem cells
o Multi-potent (produce all different cell lines and possible endothelial cells)
o Self-renewing (can maintain its own cell pool)
- Asymmetric vs symmetric
o Capable of repopulating (bone marrow transplant)
o Capable of differentiation into mature precursors
o Are present in very low numbers and are morphologically indistinct
o Capable of mobility and redistribution through the circulation
red cell maturation
o Normally 50,000 in circulation
o If have severe RBC deficiency, can lose 1g Hb/week
o Decrease in cell size
o Decreasing nuclear-cytoplasmic ratio
o Nuclear maturation (chromatin clumping and extrusion)
o Cytoplasmic maturation (hemoglobin)
erythrocyte characteristics
o Non-nucleated biconcave disc
o Slightly smaller than normal lymphocyte nucleus
o Central pallor (1/3 cell diameter)
o Released into blood as reticulocytes
o 120 day lifespan (1/120 replaced every day = 0.8-1.0 % reticulocytes)
o main function is oxygen transport. Hemoglobins:
• Hb A (a2b2)—main adult hemoglobin (95%)
• Hb F (a2g2)—1% (but main Hb in fetuses)
• Hb A2 (a2d2)—2-3%
reticulocyte characteristics
o Newly produced red cells
o Slightly larger, diffusely basophilic cytoplasm
o Supra-vital staining of RNA-ribosomal complexes (still hemoglobin synthesis)
o Increased numbers reflect increased production. Markedly increased in hemolytic processes

erythropoietin
• Erythropoietin (EPO) is main regulator of RBC production:
o Made by interstitial cortical renal cells
o HIF (hypoxia inducible factor) regulates Epo txn
o Clinical uses:
• Anemia of renal failure
• Anemia of prematurity
• Myelodysplasia (refractory anemia, sideroblastic anemia)
• Anemia of chronic disease (inflammatory or malignant)
• w/ surgical procedures (autologous transfusion)
Lab values: RBC
• RBC – Total number
o Reported as number of cells per liter of blood
o Adult male 4.5-6 x1012/L, adult female 4-5.5 x1012/L
Lab values Hemoglobin
• Hemoglobin (Hb) – Concentration
o Measured as gram per deciliter of blood
o Adult male 14-18g/dL, Adult female 12-16g/dL
o Anemia: decreased Hb
o Polycythemia: increased Hb
Hematocrit Lab values
• Hematocrit (Hct) – Volume
o Volume of red blood cells to volume of whole blood cells
o Calculated from RBC and MCV: Hematocrit = RBC (cells/liter) X MCV (liter/cell)
o Adult male 40%-54%, adult female 35%-47%
Mean copuscular volume (MCV)
• Mean corpuscular volume (MCV)
o Determined as mean of red blood cell distribution histogram, Normal range: 82-100 um3
o Microcytosis: decreased MCV
o Macrocytosis: increased MCV
MCH lab value
• Mean corpuscular hemoglobin (MCH)
o Hemoglobin concentration per cell
o Normal range: 27-34 pg
o Hemoglobin divided by RBC
o Limited clinical use
MCHC lab values
• Mean corpuscular hemoglobin concentration (MCHC)
o Average hemoglobin concentration per total red blood cell volume, Normal range 32-36%
o Hemoglobin divided by hematocrit
o Limited clinical use
RDW lab values
• RDW: Red cell distribution width:
o Coefficient of variation of red cell histogram distribution curve
o Measure degree of variation of red blood cell size (or anisocytosis)
o Normal range: 11-15%
o Increase of RDW is associated with anemia from various deficiencies: Iron, B12, folate
o Normal or low RDW is associated with thalassemia or anemia of chronic disease
o Not specific, must be interpreted in conjunction of other CBC and red cell indices
lab values reticulocytes
• Reticulocytes:
o Immature red blood cells containing residual ribosomes
o Indicator of red cell production
o Normal range: 0.5-1.5% (20-76 B/L)
o Clinically used to evaluate anemia
• Low reticulocyte count: iron deficiency, folate/B12 deficiency, bone marrow failure
• High reticulocyte count: acute blood loss, hemolysis
ESR lab values
o Measures distance of red blood cells fall in a vertical tube over a given period of time
o Normal range: 0-15 mm/hr
o Negative charges on red blood cells prevent stacking
o Inflammatory proteins (such as fibrinogen, a-, b-, g-globins) increase red cell sedimentation.
o A more rapid fall of red cells in the test tube, resulting higher stack of red cells – elevated ESR
o Elevated ESR indicates inflammatory process
• useful in monitor disease process, esp. temporal arteritis, polymyalgia rheumatica
o Not recommended for screening test or diagnostic purpose
o False positive and false negative common
MCV low, RDW normal
thalassemia trait
MCV low, RDW high
iron deficiency
MCV normal, RDW normal
chronic disease
MCV normal, RDW high
homozygous hemoglobinopathy
MCV high, RDW normal
aplastic anemia
MCV high, RDW high
folate and B12 deficiency
iron deficiency anemia lab characteristics

o Microcytic hypochromic anemia: reduced hemoglobin, reduced MCV, increased RDW
o Serum iron profile: reduced iron, reduced ferritin, increased total iron binding capacity
o Normal to low reticulocytes count, lack of polychromasia on blood smear
o Microcytic hypochromic red blood cells in blood smear: smaller red cells with increased central pallor
anisocytosis
variation of cell size
poikilocytosis
variation of cell shape
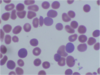
Polychromasia – Increase of reticulocytes

Spherocytes – Smaller, round shaped red blood cells which lack central pallor.
- Acquired immune hemolytic anemia
- Post transfusion
- Hemolytic anemia due to oxidant drugs
- Hemolysis due to a large spleen
- Hereditary spherocytosis
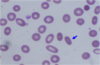
Elliptocyte (aka ovalocyte) - an elongated red blood cell with blunt end
Shape varies from slightly oval or egg-shaped to long pencil-like.
- Hereditary elliptocytosis
- Smaller number can be seen in iron deficiency, thalassemia, hemoglobinopathy, and other anemia.
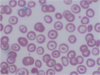
Target cell - A dense central area surrounded by a relatively clear area and a peripheral rim of hemoglobin
Thalassemia
Sickle cell disease (esp. hemoglobin C disease)
Liver disease
Post splenectomy
Iron deficiency

Sickle cell – Sickle shaped red cells, pointed at both ends, caused by molecular aggregation of hemoglobin S
Sickle cell disease, not present in sickle cell trait
Caused by a point mutation in b-globin chain
Mutated b-globin polymerizes with low oxygen
Cause changes of red cell shape

Echinocyte (Burr cells) – short, evenly space spicules and preserved central pallor
Uremia
Bleeding ulcer
Gastric cancer
Artifact
Distinguish from Acanthocyte (Spur cell) mostly seen in lipoproteinemia

Schistocyte – Distorted, fragmented cells with 2 to 3 pointed ends
Microangiopathic hemolytic anemia (DIC, TTP)
Severe burns
Prosthetic heart valves
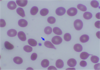
Teardrop cells – Distorted, drop-shaped cell
Myelophthisis – bone marrow fibrosis caused by various etiologies such as primary myelofibrosis, metastatic carcinoma etc.
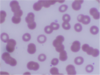
Rouleaux – cell aggregates resembling stack of coins, caused by increased paraprotein in serum
Paraproteinemia – monoclonal or polyclonal gammopathy
Artifact – thick smear

Agglutination – Cell clumping
Cold agglutinin disease
Mostly IgM againt I/i antigens on red cells
No reactive in body temperature, maximum reactivity at 4°C
May cause extravascular or intravascular hemolysis
Also artifact
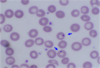
Howell-Jolly bodies – Small discrete basophilic dense inclusions usually single, nuclear remnants
Post splenectomy
Hemolytic anemia
Megaloblastic anemia

Basophilic stippling – Punctate basophilic inclusions, precipitated ribosome RNA
Various anemia – fine stippling
Thalassemia – coarse stippling
Lead intoxication – coarse stippling
microangiopathic hemolytic anemia

Include thrombotic thrombocytopenic purpura (TTP), disseminated intravascular coagulation (DIC), hemolytic uremic syndrome, uremia with hypertension, sickle cell anemia with pulmonary emboli.
Red blood cells are fragmented by intravascular fibrin deposit in TTP and DIC
Red cell morphology includes helmet, burr, acanthocyte, spur, spiculated, fragmented, pinched etc.

50-65% of leukocytes
Segmented nucleus (3-4 lobes)
Granular, pale pink cytoplasm
Circulate only briefly (12 hours)
Released into blood at band stage
Recruited into tissues (acute inflammation)
neutrophilia
Absolute neutrophil count > 7,000/ml
- *Etiology:**
1) Infectious diseases (especially bacterial)
2) Acute stress (trauma, recent surgery)
3) Acute tissue necrosis (acute MI)
4) Medications (steroids, lithium, growth factors)
5) Pregnancy (third trimester)
6) Underlying malignancy (tumor products)
Pathophysiology: Increased mobilization of neutrophils from
1) Bone marrow storage pool
2) Marginal pool of circulating blood
Increased bone marrow production secondary to colony stimulating factors
reactive morphologic findings
1) Left shift (increased number of bands)
2) Toxic granulation (increased primary granules)
3) Döhle bodies (blue cytoplasmic inclusions, aggregated rough ER)
4) Vacuolization

neutrophil leukemoid reaction versus chronic myelogenous leukemia
1) Stages of myeloid cells present (reactive has more mature cells)
2) Alkaline phosphatase activity (present in reactive)
3) Morphologic findings (toxic changes) (present in reactive)
4) Basophilia (present in CML, NOT IN REACTIVE)
5) Philadelphia chromosome (BCR-ABL)–CML
Neutropenia
- Absolute neutrophil count < 1800/ml
- Increased susceptibility to infection as neutrophil count drops below 1000/ml
- Agranulocytosis - virtual absence of neutrophils (depletion of blood and marrow storage pools)
- May need to use antibiotic prophylaxis
Pathophysiology:
- Decreased marrow production (aplastic anemia, viral suppression, drug-related, Kostmann syndrome, cyclic neutropenia)
- Ineffective marrow production (megaloblastic anemia, myelodysplasia)
- Increased peripheral destruction (antibody mediated, overwhelming infection, hypersplenism)
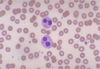
Pelger-Huët anomaly: hyposegmented neutrophils
inherited (autosomal dominant)
- acquired (myelodysplasia)

hypersegmented neutrophils: >5 segments
megaloblastic anemia, hydroxyurea
lymphocytes
25-40% of leukocytes (higher in children)
Round/oval non-segmented nucleus
Scant basophilic cytoplasm
80% are T cells (CD4:CD8 = 2:1)
Large granular lymphocytes (cytotoxic T cells, NK cells)
Function in humoral and cell-mediated immunity
lymphocytosis

Absolute lymphocyte count > 5000/ml (>7000/ml - children, >9,000/ml - infants)
Etiology:
1) Infectious diseases (especially viral)
2) Lymphoproliferative disorders
3) Immunologic reactions (drugs, serum sickness)
Types
Small mature lymphocytes (pertussis)
Reactive “atypical” lymphocytes (EBV)
1) Increased size, smudgy chromatin, may have nucleoli, abundant basophilic cytoplasm
2) Spectrum of atypical cells (CD8 T cells)
Large granular lymphocytes (HIV, rheumatoid arthritis, clonal proliferations)
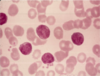

Monocytes
- 5-12% of leukocytes
- Irregular non-segmented nucleus
- Abundant blue-gray cytoplasm with some granules and vacuolization
- Migrate into tissues, become macrophages
- Function in acute and chronic inflammation
Monocytosis

Absolute monocyte count > 800/ml
Etiology:
1) Chronic inflammatory disorders
2) Chronic infectious diseases (TB)
3) Associated with neutropenia (relative)
4) Clonal disorders (monocytic leukemias)

Eosinophils
- 3% of leukocytes
- Segmented nucleus (2 lobes)
- Numerous orange-red cytoplasmic granules (basic proteins)
- Migrate into tissues (mucosal surfaces)
- Function in allergic reactions, parasitic infections
eosinophilia
Absolute eosinophil count > 350/ml
Etiology (specific growth factors - IL-5):
1) Infectious diseases (tissue parasites)
2) Allergic reactions
3) Asthma
4) Collagen vascular diseases
5) Neoplastic processes
Hypereosinophilic Syndrome

- Persistent eosinophilia (> six months) with no apparent underlying cause
- Eosinophil count often > 1500/ml
- Eosinophils have abnormal morphology
- Tissue infiltration (heart, lungs, CNS)
- Treat with steroids and/or chemotherapy

Basophil
- Up to 1% of leukocytes
- Segmented nucleus
- Numerous purple cytoplasmic granules (inflammatory mediators, e.g. histamine)
- Distinct cell from mast cell (tissue cell)
- Immediate type hypersensitivity
chronic granulomatous disease
functional leukocyte defect
1) X-linked deficiency of NADPH oxidase
2) Impaired respiratory burst and H2O2 production
3) Recurrent bacterial infections (especially catalase-positive organisms)
microcytic anemia
causes
clinical work-up
o Iron deficiencies
o Hemoglobinopathies
• Thalassemia (α-thalassemia, β-thalassemia)
• Sickle cell disease
o Membrane defects
• Hereditary spherocytosis
o Work up:
• History and physical exam
• Overt bleeding
• Symptoms of anemia
• Symptoms of systemic disease
• Labs
• CBCD and smear
• Iron studies: Fe, TIBC, ferritin
• Hemoglobin electrophoresis (Genetic studies for α thalassemia)
characteristics of iron deficiency anemia
microcytic
o Bleeding
o Cigar shaped cellso
Hypochromic
o MCV usually 70s
o May be symptomatic or asymptomatic
o Platelet count may be high.
characteristics of sickle cell disease causing anemia
• Sickle Cell Disease (microcytic)
o Associated with joint pain
o Sickle shaped cells
o Usually MCV 70s or low 80s
o Not hypochromic
anemia d/t red cell memrbane abnormalities
• Membrane abnormalities (microcytic anemia)
o Hereditary spherocytosis
• Small cells without central pallor
• MCV <70
• May have large RDW
o Hereditary elliptocytosis
o Stomatocytosis
anemia due to thalassemias
• Thalassemia (microcytic anemia)
o Usually very low MCV
o Hypochromic
o Range of symptoms from completely asymptomatic to transfusion dependent
o May have normal RBC
anemia due to B12 deficiency
macrocytic
o Associated with macroglossia, neuropathies
o May have very low h/h
o Usually indicates absorption issue
o May present with multilineage cytopenias and hemolysis
anemia due to myelodysplasia
macrocytic
o Usually presents in middle aged or older patient
o Normal or high B12 and folate
o Usually slow onset
o Bone marrow biopsy needed to diagnose
anemia of chronic inflammation/kidney disease
o Renal insufficiency or inflammation can lead to normocytic normochromic anemia
o May be macro- or microcytic
o Exogenous erythropoietin can be given with caution (Thrombotic risk!!)
o Consider whether treatment is needed/helpful
o Poor prognostic sign (esp in older patients) but not clear that treatment is helpful in the absence of symptoms
anemia d/t bone marrow infiltration
o Hematologic malignancy
• Leukemia
• Multiple myeloma
• Lymphoma
o Solid tumor
o Scar tissue
• Myelofibrosis (primary or secondary)
• HIV
o Aplastic anemia (primary or secondary)
o Pure red cell aplasia (parvovirus)
anemia d/t meds or drugs
o Can be d/t basically any med
o Hemolysis (G6PD deficiency—malarials, sulfa drugs, nitrates; lidocaine)
o Underproduction:
• Chemo (methotrexate, cyclosporine, hydroxyurea)
• OTC, supplements
anemia due to parvovirus
o Hypoplastic anemia
o Pure red cell aplasia
pt presents with fatigue, dyspnea on exertion, and dizziness on standing
he is found to have a microcytic, hypochromic anemia
He also reports cravings for ice.
on PE he is tachycardic, tachypnic, has othrostasis and appears pale
labs show dec ferritin
DX: iron deficiency anemia
Tx: oral replacement for those who can tolerate (constipation and nausea are side effects); otherwise IV replacement (anaphylaxis)
60yo woman presents with fatigue, SOB, and syncope. She also reports decreased sensation in feet.
PE: chelitis around edges of mouth, glossitis
Lab: macrocytic, megaloblastic anemia; elevated homocysteine and MMA; low–normal B12 and normal Folate
Dx:
Tx?
causes?
Dx: anemia d/t B12 deficiency
Tx: IV replacement therapy followed by oral replacement
Causes: vegan, malabsorption (esp. pernicious anemia, lack of terminal ileum etc.)
Pt presents w/ SOB, fatigue, and chest pain
PE: conjutiva are pale, tachycardic, tachypnic, orthostasis
Lab: macrocytic megaloblastic anemia, elevated homocysteine;
Dx?
Tx?
Causes?
dx: anemia d/t folate deficiency
Tx: folate replacement
Causes: inadequate intake (vegans are fine), malabsorption, alcohol, IBD, bowel resection, amyloid, scleroderma
inc. loss: CHF, dialysis, severe liver disease
hemagglutinin
IgM vs IgG
o antibody causes RBC aggregation
• forms basis for blood bank testing
o IgM antibodies:
• large pentamer is big enough to overcome repellant forces between RBCs
o IgG antibodies:
• CANNOT cause hemaglutination: its not big enough
• AHG = anti human globulin
- = blood bank laboratory reagent
- reacts with IgG antibody on RBCs → hemaglutination
- AHG bridges the gap between IgG antibodies
- allows blood bank to detect RBCs coated with IgG antibodies
direct antiglobulin test (DAT)
- determines what is on RBCs
- = DAT or direct coomb’s test
- detects IgG or C3 on RBC (C3 = footprint of IgM)
- used to determine immune hemolysis by in vivo red cell sensitization
- autoimmune hemolytic anemia, hemolytic disease of newborn, drug induced hemolytic anemia, transfusion reactions
indirect antiglobulin test (IAT)
- determines what is in serum
- = IAT or indirect coomb’s test
- detects IgG in serum
- used to determine RBC compatibility prior to transfusion
mechanism of intravascular hemolysis
• intravascular hemolysis: usually due to IgM antibodies
o cause hemaglutination in circulation
o IgM cause mechanical destruction of RBCs + complement fixation/lysis → RBC lysis causes free RBC stroma release → free RBC stroma stimulates vasoactive peptide + clotting cascades + anaphylatoxins release
o symptoms:
• back pain
• hemoglobinemia: red plasma
• hemoglobinuria: red urine
(these are not seen in extravascular hemolysis)
• fever; coagulopathy; hTN; pulmonary compromise → DIC, vascular collapse, renal failure → death
mechanism of extravascular hemolysis
• extravascular hemolysis: usually due to IgG antibodies
o no lysis of RBCs because IgG is inefficient at complement mediated lysis
o IgG coats RBCs → allows faster clearance by reticuloendothelial system
o symptoms:
• paucity of signs and symptoms
• low grade fever
• hallmark = drop in RBC count due immune destruction of RBCs
Lab findings in intra and extravascular hemolysis
o spherocytes
o circulating nucleated RBCs
o reticulocytosis
o ↑ LDH; ↑ bilirubin; ↓ haptoglobin
ABO antibodies
o IgM antibodies (some IgG can be made)
o arise naturally in individuals lacking corresponding antigen
o arise after infancy after stimulation by cross reacting environmental antigens (bacteria colonizing gut)
• (NOT inherited!)
o cause hemaglutination of antigen positive RBCs @ body T
• ABO incompatible RBC transfusion → fatal complication
• sold organ transplant rejection
Rh antibodies
o do not occur naturally, usually IgG
o alloimmunized to Rh antigens by exposure to RBC
o Rh negative patient may make IgG anti-D antibodies
• following:
- transfusion with D positive blood
- pregnancy with an Rh positive fetus
• occurs in 80% of normal, Rh negative patients when exposed to D positive RBCs
Rh factor
D antigen
D antigen + is Rh positive
D antigen - is Rh negative
Rh negative mother pregnant with and Rh positive fetus. This is her second pregnancy.
What are the potential complications?
How should she be treated?
complications: erythroblastosis, hydrops fetalis (hemolysis, anemia, hyrops, fetal demise)
Prevention:
-Rh (-) women sensitized during pregnancy with Rh (+) fetus → fetal maternal hemorrhage (FMH) during delivery → all subsequent pregnancies with Rh (+) fetuses would be at risk
• anti-D from sensitized donor plasma
- → injected into Rh (-) mothers
- RhIG = Rh immune globulin
- ↓ HDN incidence of Rh incompatible pregnancies
- only effective if Rh(-) mother is naïve to D-antigen
- ( alloimmunized Rh (-) mothers gain no benefit from RhIG)
- give @ 3rd trimester and at delivery to prevent FMH alloimmunization
ABO hemolytic didsease of newborns
o cause by caused by antibodies to other Rh antigens, the ABO system, other antigens
o milder than anti-D HDN, can occur in first pregnancy
o seen if IgG component of maternal ABO antibodies is present
• usually group O mother and group A or B fetus
o no method for prevention
basic principles of ABO compatability for RBC transfusion vs platelet transfusion
- RBC transfusion: avoid incompatibility with patients ABO hemagglutinin
- plasma transfusion: avoid incompatibility with patients antigens
basic principles for Rh compatibility
• Rh (+) can get either Rh (+) or (-) blood
• Rh (-) should only get Rh (-)
- if supply is low, reserve for females of childbearing age
- ignore Rh for plasma transfusion
- honor Rh for platelet transfusion
- platelets produce some RBCs in them
- platelet production may often be ABO incompatible but generally accepted as safe
Blood products are tested for…
o anti-HIV 1 and 2( lowest risk of transfusion transmission(1/ 2 million))
o HbsAg
o anti-HBc
o anti-HCV
o anti-HTLV 1 and 2
o syphilis
o nucleic acid testing: HIV, HBV, HCV, west Nile virus
o antibodies to trypanosomes, Chagas disease
**immunocompromised pts: must test for CMV or provide leukocyte reduced preparation
Adverse reactions to transfusions
- Acute hemolytic transfusion reaction
- delayed hemolytic transfusion reaction: pt antibodies did not show up at time of screening
- delayed serologic transfusion reaction: pts develop new antibody after transfusion
- Febrile, Non-hemolytic transfusion reaction: pt has anti-leukocyte antibodies to donor leukocytes; 1% transfusions; rigors
- Transfusion associated circulatory overload: excessive rate/volume of transfusions, CV disease, overload system
- Acute hypotensive reaction: pts taking ACE-I
- Transfusion related acute lung injury (TRALI): donor has anti-leukocyte antibodies that react w/ pt leukocytes; acute lung injury
- Transfusion associated graft vs host disease: immunocompromised pts, very severe
characteristics of hemolytic anemia (3)
- decreased RBC life span
- membrane damage
- Hb release (intravascular or extravascular)
clinical signs and symptoms of hemolytic anemia
o general symptoms of anemia
• pallor, ↓ exercise tolerance, fatigue, palpitations, dyspnea
• compensated anemia: no anemia unless complications of hemolytic anemia
o specific:
• jaundice: icterus
• dark colored urine
o splenomegaly +/- hepatomegaly seen in
• congenital chronic hemolytic anemias
- hereditary spherocytosis
- PK deficiency
- thalassemia
• acquired hemolytic anemia
- autoimmune hemolytic anemia
Lab signs of hemolytic anemia
o reticulocytosis = polychromasia
o unconjugated hyperbilirubinemia
o ↑ fecal and urine urobilinogen
o ↓ serum haptoglobin
o ↑ LDH (lactate dehydrogenase)
o ↑ AST (amino transferase)
o hemoglobinemia
- • hemoglobinuria
- • hemosiderinuria
o ↓ survival of autologous RBC labeled with Cr
Signs of chronic hemolysis
o cholelithiasis: brown bilirubin gallstones
o leg ulcers
• especially in sickle cell anemia and hereditary spherocytosis
o aplastic crises
• precipitated by infection: parvovirus B19
o hyperhemolysis
• precipitated by infection
o skeletal abnormalities
• characteristic of severe thalassemia major and sickle cell disorders
• hair on end appearance of x ray @ skull
• jaw and dental abnormalities
types of erythrocytes in hemolytic anemia (3)
discocyte: SA:V ratio>1 (normal)
Spherocyte: SA:V ratio is decreased (lose membrane)
Target cell: SA:V ratio is increased (lipid bilayer expands)
classifying hemolytic anemias
o intrinsic membrane disorders
• hereditary
hereditary spherocytosis = HS
hereditary elliptocytosis = HE
• acquired
paroxysmal nocturnal hemoglobinuria = PNH
o extrinsic membrane disorders
• cytoplasmic disorders
enzymopathies: G6PD deficiency, d/o glycolytic pathway
• defect in structure = hemoglobinopathies
o sickle cell syndrome
• defect in synthesis = thalassemia
o a thalassemia
o B thalassemia
• extracellular disorders
auto immune hemolytic anemia = AIA
infection
acanthocytosis
fragmented syndromes: microangiopathies
physical agents
other disorders
major hormone involved in platelet production
Thrombopoeitin (TPO)
made in liver
negative feedback via megakaryocytes and platelet mass
physiology of platelet adhesion
1) Tethering and rolling:
- platelet receptor: GPIb-IX-V
- ligand: vWF
2) Activation and Adhesion:
- platelet receptor: GPIa-IIa and GPVI
- Ligand: collagen
3) Aggregation:
- platelet receptor: GPIIb-IIIa
- ligand: fibrinogen, vWF
Clinical features
platelet defect vs coagulopathy
*

typical clinical presentation of platelet defect
petechiae usually on lower extremities
mucocutaneous bleeding (gingiva, epistaxis, menorrhagia)
F>M
most common causes of thrombocytopenia
- drug induced (heparin)
- pregnancy (benign, resolves after delivery)
- hypersplenism (any cause sequesters platelets)
- infection (dec. production and inc. destruction; viral (HCV, HIV), rocky mtn spotted fever, bacterial sepsis)
lab evaluation of thrombocytopenia
blood smear: platelet number, size, clumping, granularity; RBC schistocytes/spherocytes
CBC with mean platelet volume
Immature platelet fraction (IPF); reticulocytes
PT, aPTT
D-dimer
BUN/creatinine
Consider: HCV serology, PF4-heparin antibodies, antiphospholipid antibodies, abdominal CT/ultrasound, bone marrow exam
treatment of thrombocytopenia
correct underlying d/o
platelet transfusions:
- for bleeding, transfuse until platelets >40,000 and bleeding stops
- prophylactic: <10,000 (minor sugery 50,000, major surgery 80,000)
drug induced thrombocytopenia
immune mediated: heparin! etc
suppressed platlet production: chemo, ETOH, thiazides
not assoc. w/ abnormalities in other blood cells or splenomegaly
platelets recovery after drug is removed
tx: withdraw offending drug
heparin induced thrombocytopenia
1-3% heparin exposed pts
thrombocytop;enia and life/limb-threatening thrombosis
pathogenesis:
- HIT antibodies (against heparin + PF$ complex)
- PF4 (from alpha granules)
- heparin
- platelet Fc receptor (induces platelet activation, platelet consumption, and hypercoaguable state)
Clinical:
- thrombocytopenia (>50% dec from baseline)
- timing (5-10 days from heparin or <1 d if recent heparin)
- thrombosis or skin necrosis
- other causes thrombocytopenia are not present
Labs:
- PF4 ELISA: tests for presence of HIT antibodies (high sensitivity)
- Serotonin Release Assay (SRA); tests for functional HIT Ab (high specificity)
Tx: discontinue heparin and use direct thrombin inhibitors to anticoagulate
Immune (Idiopathic) thrombocytopenia Purpura
not assoc. w other abnormalities in blood cells, coagulation, or splenomegaly
adequate bone marrow megakaryocytes
autoantibody formed against platelet antigens–>autimmune peripheral platelet destruction and inadequate megakaryocyte response
Idiopathic in adults; and assoc. with viral illness in kids
Tx: corticosteroids or IV Ig
Thrombotic Thrombocytopenic Purpura (TTP)
systemic microvascular thrombosis:
- thrombocytopenia d/t platelet consumption
- microangiopathic hemolytic anemia
- tissue ischemia and infarction
d/t ADAMTS13 deficiency or inhibition–>ultralarge vWF multimers–>spontaneously thrombose
fatal if untreated
Tx: plasma EXCHANGE for acquired form and plasma transfusion for inherited form; supportive care
hereditary qualitative platelet defects
glanzmann thrombasthenia
bernard soulier syndrome
platelet storage pool defects
acquired qualitative platelet defects
Drugs:
- aspirin (lasts 7 days d/t irreversible COX-1 inhib)
- NSAIDS (reversible COX-1 inhib)
- clopidogrel (impair platelet aggregation, block ADP-% (PY212)
Uremia: uremic plasma causes inhib or normal platets (Tx to correct uremia: dialysis, correct anemia, EPO, DDAVP, crytoprecipitate, estrogen)
myeloproliferative d/o
cardiopulmonary bypass
acquired vWD
acquired storage pool disease
normal red blood cell
structure and destruction
lifespan is 120 d
RBCs removed extrvascularly by macrophages of RES (bone marrow, spleen, liver, LN)
structure: lipid and integral protein bilayer attached to cytoskeleton (spectrin)
characteristics of hemolytic anemia (3)
dec RBC lifespan
membrane damage
Hb release: intravascular or extravascular
clinical signs and symptoms of hemolytic anemia
gen sx of anemia: pallor, dec exercise tolerance, fatigue, palpitations, dyspnea
compensated anemia may be asymptomatic
complications of hemolysis–>jaundice, dark colored urine
splenomegatly +/- hepatomegaly (hereditary spherocytosis, PK deficiency, thalassemia, autoimmune hemolytic anemia)
lab signs of hemolytic anemia
reticulocytosis=polychromasia of RBC
unconjugated hyperbilirubinemia
dec. serum haptoglobin
inc. LDH
hemoglobinemia (hemoglobinuria, hemosiderinuria)
signs of chronic hemolysis
cholelithiasis: brown pigment gallstones
leg ulcers (esp. sickle cell anemia, hereditary spherocytosis)
aplastic crisis (ppt’ed by infection esp. parvovirus)
hyperhemolysis (ppt’d by infection)
skeletal abnormalities (severe thalassemia major, sickle cell d/o; hair on end appearance on X-ray, jaw and dental abnormalities)
3 types of erythrocytes
discocyte: SA:V ratio >1
spheroctyes: dec. SA:V ratio (lose membrane)
target cell: inc. SA:V ratio (gain membrane)
hemolytic anemias d/t intrinsic membrane d/o
hereditary:
- hereditary spherocytosis
- hereditary elliptocytosis
Acquired: paroxysmal nocturnal hemoglobinuria
hemolytic anemias d/t extrinsic membrane abnormalities
G6PD deficiency
d/o of glycolytic pathway
sickle cell syndrome
alpha and beta thalassemia
autoimmune hemolytic anemia
hereditary spherocytosis
d/t spectrin (cytoskeletal protein) deficiency
pathophysiology
- membrane loss: dec. SA:V ratio=spherocytosis, inc. osmotic fragility, dec RBC deformability
- splenic trapping
- hemolysis (mostly extracellular)
Clinical
- chronic anemia: pallor, jaundice, dark colored urine, splenomegatly, cholelithiasis, chronic leg ulcers
- crisis: aplastic, hyperhemolytic–assoc. w/ parvovirus infection; leukopenia/thrombocytopenia, 7-14d, may require folic acid suppl
Labs:
- spherocytosis, reticulocytosis
- hyperchromia d/t inc. Hb conc
- anisocytosis w/o poikilocytosis
- inc. osmotic fragility, dec. RBC deformability
Tx:
- symptomatic
- splenectomy (cures disease, use in moderate–severe cases, vaccinate encapsulated organisms)
hereditary elliptocytosis
most is mild and not clinicall sig
hereditary pyropoikilocytosis (HPP)
- rare, homozygous form, AR
- severe anemia
- microspherocytes, poikilocytes, splenomegaly
- RBC fragmentation, heat sensitivity
parosysmal nocturnal hemoglobinuria
only acquired intrinsic membrane abnormality
deficiency of GPI–>dec complement regulators CD55 and CD59–>sensitivity to complement and intravascular lysis
mutation in PIG-A gene on X-chromosome (more common in females)
assoc. w/ aplastic anemia, venous thrombosis, pancytopenia, iron deficiency (may manifest as budd-chiari syndrome)
Dx by flow cytometry
Tx: symptomatic or eculizumab
G6PD deficiency
G6PD is only source of NADPH in RBC
sex linked=more common in males
type B: western, provoked by oxidative stress, more severe; fava bean sensitivity
Type A: africans and AAs;
- provoked by oxidative stress–>hemolysis
- fever, drugs, infection
- anemia is not seen unless RBC oxidant stress exposure: primaquine, dapsone, nitrofuranotoin, acidosis, infection, fava bean sensitivity
Screening:
- NADPH+blue dye=colorless if enzyme is present
Clinical features:
- acute intravascular hemolysis
- hemoglobinemia, hemoglobinuria, jaundice within 1-3days of giving drug
- mild hemolysis
- commonly acute anemia sx
- severe cases: abdominal or back pain
- heinz bodies on blood smear
pyruvate kinase deficiency
d/o of glycolytic pathway=dec. ATP production and inc. cation permeability
AR
chronic hemolysis
splenomegaly
macroovalocytosis
inc. 2,3 DPG (dec. adverse effects b/c inc. O2 release from Hbg)
autoimmune hemolytic anemia
acquired d/o in which autoantibodies against RBC
clinical:
- anemia of variable severity
- splenomegaly
- positive direct coombs test (AHG test)
Dx based on autoantibodies (IgG) or complement (C3d or C4) attached to RBC
Warm AIHA:
- middle aged women
- IgG mediated (splenic clearance, C amplifies effect)
- responds to prednisone and/or splenectomy
- coombs test: +IgG; +/- C
Cold AIHA:
- IgM mediated (hepatic clearance, C dependence)
- does not respond to prednisone/splenectomy–>keep pt warm
- coombs test: + for C only
coombs test
direct: positive test indicates presence of Ab on RBC
indirect: postiive test indicates presence of Ab in serum

alpha thalassemia
4 alpha genes:
- 1 missing: silent carrier
- 2 missing: alpha-thalassemia trait
- 3 missing: Hemoglobin H disease
- 4 missing=hydrops fetalis, death in perinatal period
Hemoglobin H disease:
- absence of 3 alpha Hb genes–>little a-globin–>formation of b-globin tetramers (HbH)
- HbH is unstable and easily oxidized–>forms inclusions in RBCs–>hemolysis
- clinical disease: moderate anemia, hemolysis, sensitivity to oxidative stress
- Tx: transfusion, splenectomy, iron chelation
Hemoglobin Barts: fetus/newborn w/ any alpha-thalassemia forms tetramers of gamma-chains (stable but poor oxygen release)
beta-thalassemia trait/minor
single gene mutation
mild asymptomatic anemia
beta-thalassemia intermedia
homozygous thalassemia + or B thal/HbE or mixed
Hb in 5-10 range
some skeletal abnormalities
hepatosplenomegaly
beta-thalassemia major/Cooley’s anemia
absence or severe underproduction of both beta globin genes
fatal early in life if not treated with transfusion
skeletal abnormalities, hepatosplenomegaly
iron overload–>death in teens unless treated
- iron overload d/t multiple transfusions
- iron deposition=MOD (esp cardiac, liver, pituitary)
- chelation w/ desferol, deferasirox, deferiprone
lacks inflammatory markers
Tx:
- transfusions to keep Hb>9g/dl
- splenectomy (inc. lifespan RBC)
- iron chelation
- bone marrow transplant limited (sometimes in children)
sickle cell disease
pathophysiology
heterozygous=sickle trait=benign
homozygous=sickling under hypoxic conditions (severe stress, high altitude, dehydration–>sickle Hb polymerization)
sickled cells are incapable of traversing cpaillaries–>small vessel thrombi–>severe pain + organ dysfx
sickle cell disease variants
Hemoglobin C: lysine mutation instead of valine
thalassemia/sickle: looks like sickle cell anemia, cells are smaller (dec. MCV)
SC disease: sickle Hbg/C-Hbg–>milder form (cells do not sickle but dehydrate–>abnormally dense cells–>crisis like syndrome)
sickle cell/single alpha-thalassemia: milder disease d/t dec Hgb in cell–>dec chance of sickling
sickle cell Hbg/HPFH–>milder disease d/t hereditary persistance of fetal hemoglobin
sickle cell disease clinical
spontaneous cell lysis and RBC turnover
inc. thrombosis/infarction (strokes, pulmonary infarction)
chronic inflammation (vs thalassemia which has no inflammation)
secondary complications due to infarcts:
- splenic infarcts (leads to splenectomy and inc. susceptibility to infections)
- joint damage (infarcts in joints)
- non-healing skin ulcers
- retinopathy; nephropathy
- severe pain
- opiate addiction
Prognosis: getting better, but sig. mortality in infancy/childhood
Variable phenotype: HPHF, single alpha gene mutations
Tx:
- hydroxyurea: inc. HbF which doesn’t sickle; and enlarges cells which dec. Hb conc.=less sickling (not useful in SC disease)
- crisis management: pain control, IV fluids, oxygen
- folic acid
- pain meds: usually opiates
- exchange transfusion: pt w/ stroke, recurrent priapism, recurrent acute chest syndrome
- bone marrow transplant in children
acute chest syndrome
complication of sickel cell disease
preceded by pneumonia, infarction, embolus
smoking inc. risk
50% idiopathic
tx is exchange transfusion
hereditary persistence of Hemoglobin F
not pathologic
when assoc. w/ sickle cell anemia or SC disease–>diseases are milder d/t dec. sickling
homozygous hemoglobin C
asymptomatic (look like thalassemia minor)
hemoglobin E
B thal/HbgE (looks like beta thalassemia intermedia)
Hb E disorder: form of beta thalassemia
- SE asians
- mild microcytic anemia
recombinant EPO
normal epo made in kidney; rhEPO made in mammalian cells (glycosylation affects half-life)
some pts develop antibodies
EPO travels from blood to bone marrow to stimulate RBC production
clincal uses:
low Epo levels
- anemia of renal failure
Normal/High Epo levels:
- anemia of prematurity
- myelodysplasia
- myelofibrosis
- multiple myeloma
- post-chemo anemia
- anemia of chronic disease
- w/ surgical procedures
Dosing:
- dec. dose in renal pts
- response measured by inc. reticulocyte and inc. Hct
ADE:
- HTN and thrombotic phenomena
- anti-epo ab: pure red cell aplasia
- epo receptor on tumor cells
- rare allergy
role of G-CSF in hematopoeisis
made in monocytes, lymphocytes, fibroblasts, endothelial cells
stimulates granulocyte production
activates phagocytic activity of mature neutrophils
mobilized hematopoietic stem cells to circulating forms
recombinant G-CSF
filgastrim and pegfilgastrim (longer half-life)
ADE: bone pain, edema
Uses:
- treatment and prevention of neutropenia after chemotherapy
- collection of stem cells for transplant
TPO and recombinant TPO
thrombopoeitin made in liver and stimulates proliferation of megakaryocytes precursors and platelet production
high levels are made and bound to receptors on platelets/megakaryocytes
*
megaloblastic anemia
macrocytosis + hypersegmented neutrophils (>5 lobes)
causes:
- age and diet
- GI disease or surgery
- pernicious anemia
- Vit B12 deficiency
- Folate deficiency
- meds (PPIs, anticonvulsants, sulfa drugs, DNA synthesis inhib–methotrexate, hydroxyurea, anti-virals)
Clinical: glossitis (loss of papillae on tongue)
pernicious anemia
cause of megaloblastic anemia (macrocytosis + hypersegmented neutrophils)
Vit B12 deficiency due to autoimmuntiy against IF or parietal cells
Tx w/ vit B12 injection (not oral!)
Vit B12 deficiency
Due to:
- inadequate diet (vegan)
- malabsorption (pernicious anemia, partial or total gastrectomy, stagnant loop syndrome, chronic tropical sprue, ileal resection, Crohns disease, congential malabsorption with proteinuria, fish tapeworm, drugs–metformin)
Absorption: requires intrinsic factor and is primarily absorbed in ileum
B12 is stored in the liver
nromal function:
- synthesis of methionine
- cofactor of folic acid function
Deficiency–>DNA synthesis impairment
- megaloblastic anemia + neurological d/o (not seen in folate def)
- neuropathy
Tx: B12 replacement, initially IV then IM (also oral)
folic acid defiicency
normal role of folate:
- found in plants and animal sources
- increased requirements during pregnancy and hemolytic anemia
- absorbed in proximal jejunum
- storage is short (compared to B12 3-5yrs)
- Required for synthesis of methionine, DNA methylation, and DNA synthesis
Deficiency:
- megaloblastic anemia + hypersegmented neutrophils
- glossitis
- NO NEUROLOGICAL ISSUES
Tx: replacment (oral, IM, IV); luecovorin=derivative than bypasses methotrexate inhibition
iron deficiency anemia
- characterized y microcytosis/elongated pale RBCs
- iron absorption is mostly at duodenum
- most iron is stored in circulating RBCs as hemoglobin, also some in bone marrow, hepatocytes, parenchmyal cells; undergoes enterohepatic recirculation
- Inc. need for iron: infants–>adolescents, pregnancy, menstruating females
- Caues of iron deficiency:
- bleeding (GI, urinary, menstruation)
- malabsorption
- inc demand (infants, teens, pregnancy, lactation)
- poor diet
- labs: dec iron saturation and inc. TIBC; dec. ferritin
- Tx:
- oral: ferrous sulfate (DOC), ferrous gluconate, ferrous fumarate; AE: nausea constipation
- IV: inidcated w/ intolerance to oral therapy, malabsorption, massive iron loss (iron dextran); ADE: anaphylaxis
essential nutrients iron vs folate vs B12

4 components of hemostasis
blood vessels:
- constriction
- subendothelial collagen, tissue factor
Endothelial cells:
- secrete platelet inhibitors, vasodilators, plasminogen activator
- surface for anticoagulants
platelets:
- plug formation
- phospholipid for coagulation
plasma proteins:
- coagulation factors
- coagulation inhibitors
- clot dissolution: fibrinolysis
venous vs arterial thombosis
veins: low flow rate, low shear rate–>fibrin rich/platelet poor clots
Arteries: high flow rate, high shear rate–>platelet rich, fibrin poor clot
site of thrombosis dictates type of thrombolytic therapy (eg. aspirin is antiplatelet and treats arterial clots)
role of endothelium in hemostasis
=anticoagulant
• endothelium promotes blood fluidity via:
o serving as protective barrier (eparating hemostatic blood components from subendothelial factors (TF and collagen); highly negative charge → repels platelets (-) charge)
o produces platelet activation inhibitors
o produces blood coagulation inhibitors
o produces fibrinolysis promoting factors → clot dissolution
• inhibits thrombus formation:
o NO, PGI2, ADPase → inhibit platelet activation
o thrombomodulin (w/ Protein C and S) → degrades FVa and FVIIIa
o TFPI: tissue factor plasma inhibitor → inhibits TF
o heparin and antithrombin III → inhibits thrombin and FXa
o plasminogen activators
• consequences of endothelial damage/activation
o synthesis of pro-coagulants: TF, PAI1
o secretion of von willebrand’s factor
o possible up regulation of luminal adhesive molecules
o reduced anticoagulants (thrombomodulin)
role of platelets in hemostasis
1) tethering and rolling:
- vWF binds subendothelial collagen which binds GPIb on platelets
- vWF made in endothelial cells
2) adherence:
- collagen binds GPVI and GPIa-IIa
- platelets release + feedback granules (ADP, TXA2)
3) aggregation:
* fibrinogen and vWF promote aggregation by binding GPIIb-IIIa on platelets
extrinsic coagulation cascade
rapid response! principle initiating event of in vivo coagulation
TF (not normally exposed to blood); expressed on vascular adventitial cells
TF + F7–>7a:TF complex–>activates 10
measured by PT
intrinsic coagulation cascade
- charge–>activates 12–>activates 11–>activates 9–>9a:8a–>activate 10
measured by aPTT
common coagulation pathway
10a + 5a–> cleave prothrombin to thrombin–>cleaves fibrinogen to fibrin
factor 13 crosslinks fibrin monomers
platelets enhance activity of coagulation factors
- Factor 9a/8a/Ca/Platelets (enhance activation 10)
- Factor 10a/5a/Ca/Platelets (enhance activation thrombin)
phospholipid membrane provides surface to speed up rxns
TFPI
Tissue factor pathway inhibitor
made by endothelial cells
TFPI–>inactive 10a:TFPI complex–>inactivates TF:7a complex
(altogether blocks 10a and TF:7a complex)
antithrombin
made in liver
high plasma concen
serine protease inhibitor forms inactive complexes w/ 12a, 11a, 10a, 9a, 7a:TF
activity is accelerated by heparin
protein C and S
degrade factors 5 and 8
fibrinolysis
maintains vascular patency and inhibits excessive clotting
plasminogen–>plasmin–>degrades cross-linked fibrin
t-PA and u-PA enhance conversion plasminogen–>plasmin
a2-antiplasmin and PAI-1 inhibit conversion plasminogen to plasmin
PT (prothrombin time)
o reflects extrinsic and common pathway
o depends on factor VII, X, V, II +/- fibrinogen (I)
o used for
• monitoring warfarin (Coumadin) therapy
• screening test for
- vitamin K deficiency (2, 7, 9, 10)
- factor VII, X, V, II deficiency
- (2, 5, 7, 10)
- liver disease
- very rare acquired factor inhibitor (autoimmune)
o performing test
• tissue factor (TF) + plasma (factor VII, X, V, II, I) + Ca+ → fibrin clot formation
o _prolonged PT + normal aPTT → factor VII deficiency _
INR
similar to PT
designed to monitor warfarin as anticoagulant
typical target INR is 2-3
aPTT
activated partial thromboplastin time
o reflects intrinsic and common pathway
o depends on all factors except VII and XIII
o used for
• monitoring heparin therapy(heparin potentiates AT III = forms inactive complex with factors 9,11,12,10,2,F7:TF)
- screening test for**
- factor VIII, IX, XI, XII deficiencies
- hemophilia A: F8 deficiency
- hemophilia B: F9 deficiency
- prekallikrein, HMWK deficiency
- von willebrand disease
- lupus anticoagulants
- acquired factor inhibitor
mixing studies
first step in evaluating prolonged PT or aPTT
mix patient plasma w/ normal plasma
if corrects–>deficiency
if it does not correct–>inhibitor
2 principles:
- inhibitors are present in excess
- 50% of any factor is adequate to provide normal test results
TT (thrombin time)
evaluate conversion of fibrinogen to fibrin
inc. reptilase time:
- low/absent or dysfunctional fibrinogen
- high levels of fibrin split products (DIC)
- myeloma and proteins act as anithrombin
inc. fibrin time:
- all of the above
- heparin or DTI
- helps differentiate b/t a heparin induced problem or not
quantitative fibrinogen
claus method (thrombin time of dilute plasma=proportional to fibrinogen conc)
indications for test:
- evaluate prolonged PT or aPTT
- DIC or bleeding (esp. w/ liver disease)
abnormally low values:
- liver disease
- DIC or consumptive states
- thrombolytic therapy
- congenitally low/absent fibrinogen
- abnormal fibrinogen protein
abnormally high values:
- acute and chronic stable liver disease
- acute phase reactant
D-dimer test
- indicates both ongoing coagulation and fibrinolysis (thrombin +13a to generate X-linked fibrin monomers and plasmin to cleave X-linked fibrin)
- used to evaluate:
- outpt DVT, PE
- DIC
-excreted by kidneys
specific coagulation factor assays
determines extent to which plasma corrects clotting time of plasma deficient in only one clotting factor
Eg: add pt plasma to factor 8 deficient plasma–>do aPTT on mix–>compare results to standards
antiphospholipid antibody testing
antiphospholipid antibody syndrome (inc. risk of blood clots and recurrent pregnancy loss)
-testing for antibodies: anti-cardiolipin, B2-glycoprotein use ELISA
lupus anticoagulant (LA)
3 criteria:
- inc. phospholipid dependent clotting time (inc PT, aPTT, DRVVT)
- failure of correction of 1:1 mix w/ normal plasma (inhibitor)
- correction with addition of excess phospholipid
DRVVT: dilute russel viper venom time = screening test for LA
isolated prolongation of PT
Causes:
- liver disease
- mild DIC
- mild Vit K deficiency (factor 7 has short halflife and is Vit K dependent)
- warfarin
- factor 7 deficiency (very rare)
lab approach:
- repeat
- does mixing correct PT?
- if no=inhibitor
- if yes=factor assays
isolated prolongation of aPTT
No bleeding: F12, prekallikrein, HMWK
- lupus anticoagulant
- heparin
- prekallikrein deficiency
- factor 12 deficiency
- HMW kininogen deficiency
Bleeding: vWF, F 8,9,11
- vWF disease
- hemophilia A (F8 def)
- hemophilia B (F9 def)
- Hemophilia C (F11 def)
- heparin
Lab approach:
- repeat–>does mixing correct?
- if no–>work up inhibitor (LA)
- if yes–>work up vWF disease or specific factor assays
prolongation of both PT and aPTT
multiple factor deficiencies caused by:
- severe liver disease
- DIC
- coumadin (warfarin)
- severe vit K deficiency
- therpeutic fibrinolysis
- dilutional (massive transfusion)
- isolated deficiency of fibrinogen, factors 2, 5 or 10 (common pathway factors)
Lab approach:
- does mixing correct?
- if not–>inhibitor
- if yes–>TT and factor assays
bleeding time test
measure of platelet function
measure of duration of bleeding following standard incision
indications:
- screening for platelet function defects
- assessment of response to therapy (DDAVP)
inaccurate in predicting operative bleeding
inc. w/ aspirin, uremia, VWD
dec with ITP
PFA-100
platelet function analyzer
measures time for citrtated whole blood to occlude a pinpoint hole at end of tube
abnormal PFA-100:
- thrombocytopenia, anemia
- VWD, afibrinogenemia
- acquired d/o of platelet function (storage pool d/o uremia, anti-platelet drugs)
- congential d/o of platelet function (bernard soulier syndrome, glanzmann thombasthenia, storage pool d/o)
platelet transmission aggregometry
less light absorbed=less aggregation
more light absorbed= more aggregation
measures response to various agonists (ADP, epinephrine, collagen, ristocetin, thrombin)
monitoring anti-platelet therapy
therapies:
- IIb-IIIa inhibitors (abciximab, integrilin)
- aspiring (COX-1 inhibt)
- clopidogrel (P2Y12 inhib)
Assays:
- standard lumiaggregometry
- PFA-100
- flow cytometry for VASP
- verify now (aspiring, IIbIIIa inhib, clopidogrel)
Rationale:
- marked variability in inter-individual response to antiplatelet agents
- lack of inhibition correlated with recurrent coronary events and poor outcomes after PCI
- inc. drug will inc. platelet inhib
aspirin
irreversible inhibitor of COX-1 (inhibits formation thromboxane A2)
irreversible platelet inhibitory effect (lasts lifespan of platelet ~1wk)
absorbed in stomach and intestine
platelets inhibited within 1 hr (good for acute situations)
NSAIDS
reversible COX-1 inhibitor (inhibits TxA2 and platelet function)
**note: NSAIDS compete with aspirin and antagonize each others actions when given together (problematic due to NSAIDS long half-life)
dypyridamole
PDE inhibitor–>inc. intracellular cAMP–>inhibits platelets
vasodilator and antiplatelet properties
cilostazole
PDE inhibitor–>inc cAMP–>inhibits platelets
promotes vasodilation and inhibits platelet aggregation
used to treat intermittent claudication
clopidogrel (plavix)
irreversible P2Y12 inhibitor (blocks platelet ADP-R and platelet aggregation)
oral ingestion, prodrug requires CYP450 (CYP2C19) for activation
**problem sig number of people have mutation CYP2C19 and poor metabolism (test pts before giving)
activity takes 3-5days (not for acute situations)
ticlopidine
P2Y12 inhibitor (blocks ADP-R and platelet aggregation)
antiplatelet effect takes 2 weeks (not for acute situations)
does not require CYP2C19 but has significant ADE
pesugrel, ticagrelor
P2Y12 inhibitor (blocks ADP-R and platelet aggregation)
not sensitive to CYP2C19
reversible inhibition P2Y12 (vs clopidogrel is irreversible)
not currently in widespread use
abciximab
aIIbB2 integrin/GPIIb-IIIa inhibitor
administered IV, short half-life, no meds to reverse effects
monoclonal Ab
tirofiban
aIIbB2 integrin/GPIIb-IIIa inhibitor
administered IV, short half-life, no meds to reverse effects
tyrosine derivative
severe, but reversible thrombocytopenia
eptifibatide
aIIbB2 integrin/GPIIb-IIIa inhibitor
administered IV, short half-life, no meds to reverse effects
bivalirudin
direct thrombin inhibitor (blocks thrombin activation)
IV
no way to reverse drug effects
primarily used when heparin can’t be used (HIT)
antiplatelet drugs in acute coronary syndrome
first line: aspirin
2nd line:
- ADP inhibitors (clopidogrel) given as dual therapy w/ aspirin (added risk bleeding, neutropenia, thrombocytopenia)
- DTI:
antiplatelet drugs in Afib
warfarin or vitamin K
antiplatelet drugs in TIA
high dose aspirin: can dec. incidence but sig ADE (GI bleed, intracerebral hemorrhage)
**use combination therapy:
- low dose aspirin + dipyridamole (PDE inhib)
- low dose aspirin + ADP antagonist (clopidogrel)
pt w/ chest pain and coronary artery blockage, has history of heparin 36hrs previously
which antiplatelet drug?
abciximab: used to prevent thrombotic events during PCI, available IV, immediate short term actions
pt has TIA, which antiplatelet drug?
low dose aspirin + clopidogrel
or low dose aspirin + dipyridamole (PDE inhib)
female takes daily aspirin and presents with chest pain. She requires prevention for future MI. She has a CYP2C19 mutation. which antiplatelet drug?
aspirin + presugrel (doesn’t require CYP2C19 and is oral drug)
acquired hemorrhagic d/o due to liver disease
deficiency of many coagulation factors
Factor VII has shortest half-life and is most sensitive to liver failure–>prolonged PT
dec. clearance of fibrin degradation products
dec. carboxylation of vit K dependent facotrs
thrombocytopenia (2/2 congestive splenomegaly and dec. TPO (produced in liver))
Tx: supportive; can give FFP and platelet transfusion
acquired hemorrhagic d/o due to renal failure
qualitative platelet dysfunction related to degree of uremia
acquired hemorrhagic d/o due to DIC
Causes:
- excess TF (OBGYN complications, promyelocytic leukemia, malignant tumors, massive trauma)
- endothelial cell injury/factor 12 activation (septicemia w/ gram neg organisms, shock)
bleeding d/t excessive intravascular coagulation (eats up substrates); fibrinolysis always present to some degree
Lab tests: none are specific to DIC
- prolonged PT, aPTT, TT
- dec. fibrinogen
- thrombocytopenia
- fibrin degradation product
- D-dimer
microangiopathic hemolytic anemia (see schistocytes on blood smear)
Tx:
- underlying cause
- supportive care
- transfusion: FFP, cyroprecipitate, platelet
- if disease is more thombotic can use heparin
- if disease is more fibrinolytic can use antifibrinolytics
vitamin K deficiency
factors 2, 7, 9, 10 protein C and S dependent on vit K
Factor 7 has shortest half-life–>can have elevated PT compared to aPTT (if both are elevated indicates severe deficiency)
Causes:
- inadequate intake, malabsorption
- antibiotics that inhibit vit K epoxide reductase
- hemorrhagic disease of newborn (severe vit K def)
- coumadin/warfarin overdose (inhibits Vit K epoxide reductase)
Labs:
- prolonged PT
- prolonged PT and aPTT with severe deficiency
- clinical diagnosis w/ correction of PT with Vit K
Tx:
- oral or parenteral vit K
- FFP effective but not required
coumadin/warfarin overdose:
- inc. INR w/o bleeding–>withhold warfarin
- Inc. INR with bleeding–>withhold warfarin, give Vit K, consider FFP or PCCs
hemophilia A
Factor 8 deficiency (F8a:9a complex is critical in activation of Factor 10)
X-linked, affects males (mostly inherited but 1/4 are spontaneous)
factor 8 circulates as noncovalent complex with vWF
Clinical:
- severe: F8<1%; spontaneous joint/soft-tissue bleeds
- Moderate: F8 1-5%; excessive bleeding with minor trauma/surgery; spontaneous bleeding is less common
- mild: F8>5%; only bleed trauma or surgery
Dx:
- severe made within first year of life
- screening test is aPTT
- Diagnostic test is F8 levels
Tx:
- factor replacement therapy (issues with pt forming antibodies to recombinant factor)
- mild pts can use DDAVP (vasopressin analog, induces robust release of F8 from endothelium; severe forms do not respond)
hemophilia B
x-linked; males
Factor 9 deficiency (F9a:8a complex activates factor 10); F9 is vit K depedent
impossible to distinguish from hemophilia A clinically
Lab:
- screening test is aPTT
- distinguish from hemophilia A w/ factor specific assay
Tx:
- recombinant or plasma derived F9 concentrates
hemophilia C
screening test is aPTT
deficiency of Factor 11
inherited AR
ashkenazi jews
Tx is FFP
bleeding in areas of inc. fibrinolysis (oral cavity, GU tract); phenotype is highly variable
VWD and normal vWF
vWF is manufactured in endothelial cells and megakaryocytes
- stored in weibel-palade bodies in endothelium
- stored in alpha-granules in platelets
Uses:
- tethers platelets to subendothelial collagen after vascular injury
- chaperone for Factor 8
requires polymerization to be effective (highest molecular wt multimers are most hemostatic)
vWF inc. in pregnancy, OCP use, liver disease, inflammation, exercise, stress, traumatic venipuncture, post-op
baseline vWF levels vary w/ ABO blood group
VWD types
genotype is equally common in males and females; but phenotype is more common in females due to OBGYN
Type 1: partial quantitative defect (most common)
Type 2: qualitative defect
- 2A: loss of high Mw multimers
- 2B: GOF mutation + excess platelet binding=inc. clearance
- 2M: poor function with normal multimer pattern
- 2N: impaired F8 binding
Type 3: complete absence of vWF
lab diagnosis treatment of VWD
Treatment:
- DDAVP/stimate: effective in type 1 and some type 2, ineffective in type 3, absolutely contraindicated in type 2B
- plasma derived vWF-F8 concentrates
- cryoprecipitate
- antifibrinolytic therapy
- hormones for menorrhagia
Lab dx:
- abnormal PFA-100
- inc. bleeding time is good screening test
- ristocetin cofactor assay
- quantity and distribution of vWF multimers using electrophoresis
virchow’s triad of thrombus formation
- stasis
- impaired vascular integrity
- systemic hypercoagulability
4.
DVT
Clinical:
- asymptomatic
- extremity pain or swelling
- 5% die if untreated
Diagnosis:
- doppler ultrasound
- venogram
PE
Clinical:
- asymptomatic
- SOB, chest pain, tachycardia, anxiety, hemoptysis, death
Diagnosis:
- CXR
- Lung V/Q scan
- Chest CT
- pulmonary angiogram (rarely done, used to be gold standard)
- Blood D-dimer: high negative predictive value
pathogenesis of acquired venous hypercoagulable states
TF exposure
stasis
inc. coagulation
dec anticoagulation
genetic
EC dysfx
EG:
- surgery/trauma (TF exposure, stasis)
- immobilization (stasis)
- malignancy (TF exposure,stasis)
- pregnancy (stasis, inc. coag, dec anticoag)
- etc.
hypercoagulability d/t malignancy
malignancy is very potent risk factor
risk highest 0-3months after diagnosis
high on differential when no other risk factors
antiphospholipid antibody syndrom (APLS)
acquired autoimmune thrombophilic condition
manifested by vascular thrombosis or recurrent pregnancy loss
may be assoc. w/ other autoimmune conditions
clinical:
- VTE: DVT or PE
- arterial thrombosis: stroke, TIA, MI
- recurrent fetal loss
- thrombocytopenia
Dx: antiphospholipid antibodies
- functional clotting assay: lupus anticoagulant (inc. phospholipid dependent clotting time, failure to correct w/ mixing, correction w/ addition of excess phospholipid)
- antigenic assay: anti-cardiolipin antibodies or anti-B2 glycoprotein antibodies
hypercoagulability due to pregnancy
6x inc. risk VTE
PE is most common cause of maternal death
post partum risk is greater for 6 wks
mechanisms: stasis, venous compression from gravid uterus, altered hemostatic factors
hypercoagulability d/t acquired hyper-homocysteinemia
dietary deficiency of B12, B6, or folate (can’t convert homocysteine to cysteine)
3 features of thrombotic microangiopathy
- microvascular thrombosis
- microangiopathic hemolytic anemia
- thrombocytopenia
schistocytes on blood smear
DDx:
- thrombotic thrombocytopenic purpura
- Hemolytic Uremic syndrome
HUS (hemolytic uremic syndrome)
systemic thrombotic microangiopathy
renal failure predominance
sporadic in adults
assoc. with E. coli O157:H7 in children
also assoc with DIC, malignant HTN, vasculitis, SLE, APLS, HIV, renal allograft
clinical manifestations of inherited thrombophilias
50% occur w/o provocation
manifest in mid-late 20s
DVT of legs and pelvis is most common
inc. incidence of superficial thrombophlebitis
arterial thrombosis is not assoc. w/ APCR or prothrombin 20210 and is asssoc with deficiences of AT, Protein C and S
Causes:
- APC resistance
- abnormal anticoagulant (AT deficiency, protein C/S deficiency)
- excess procoagulant (prothrombin 20210A or excess factor 8 or 11)
- homocysteinemia (cystathione b-synthase or MTHFR deficiency)
- fibrinogen abnormalities (dysfibrinogenemia)
- fibrinolytic defects (plasminogen def, tPA deficiency, excess TAFI)
APC resistance
hypercoagulable state=thrombosis
most commonly due to factor 5 leiden mutation (+/- or -/-)–>protein C is unable to inactivate F5
hypercoagulability d/t excess procoagulant
prothrombin 20210 mutant (+/- or -/-)
- inc. prothrombin levels
- high prevalence in northern europeans
elevated factor 8
- inherited or acquired, high prevalance VTE
elevated factor 11
antithrombin deficiency
AT normally inactivates thrombin, Factor 12, 11, 9, 10 (heparin potentiates action)
VTE in young adults
heterozygotes
may be heparin resistance
thrombosis during pregnancy is very common
protein C and protein S deficiency
hypercoagulable state presents as VTE esp in young adults
heterozygotes (homozygous= fatal neonatal purpura fulminans)
prone to warfarin induced skin necrosis
inherited homocysteinemia
hypercoagulable
measure homocysteine levels
mutations in cystathione b-synthase of MTHFR
argatroban
direct thrombin inhibitor
indication: anticoagulant for prophylaxis, thrombosis treatment, or PCI in HIT patients
works at active site of thrombin
hepatic elimination (half-life inc. w/ hepatic impairment)
IV infusion with rapid effect
no known antidote (risk of bleeding)
Lepirudin
direct thrombin inhibitor
first line treatment of HIT
bivalent inhibitor of thrombin
IV infusion w/ rapid therapeutic effect
renal excretion
no know antidote (risk of bleeding)
DTI benefits and Risks
benefits:
- independent of antithrombin
- inhibits clot bound thrombin
- lack of interaction with HIT antibody
- predictable dose response curve
- short half-life
- rapid therapeutic effect
- easily monitored
- used in cardiology and HIT
Problems:
- no reversal agents
- does not effect thrombin generation
- can have rebound activation of coagulation after discontinuation of drug
- narrow therapeutic window in cardiology
- expensive
HIT diagnosis and management
- HIT can occur in any pt with heparin exposure (usually 4-14 days post-exposure in naive pts, or immediate if recent heparin exposure)
- results in severe life threatening TECs
- consider HIT when platelets decrease 50% from baseline or <150,000 or if TEC develops
- tx: discontinue all forms of heparin immediately and treat w/ DTI (don’t delay treatment with alternative anticoagulant)
- don’t use warfarin in acute HIT
fibrinolytic drugs
plasminogen activators=clot busters
tPA or uPA (activate plasminogen–>plasmin)
plasmin degrades X-linked fibrin
ADE: hemorrhage
Inhibitors of fibrinolysis
plasminogen activator inhibition (PAI-1)
a2-antiplasmin
thrombin activatable fibrinolysis inhibitor (TAFI)
anticoagulation for DVTs and PE
goals of tx:
- prevent propogation of clot
- prevent recurrent DVT and PE
treat w/
- UFH (achieve therapeutic aPTT in first 24hr)
- LMHW (dose by actual body wt and assess renal fx)
- start anticoagulant therapy w/ warfarin in first 24h
unfractionated heparin (UFH)
binds AT–>inhibits 10a and thrombin
elimination by reticuloendothelial system
reversing agent is protamine sulfate
monitor w/ aPTT (prevent recurrent DVT, PE; reduce risk of bleeding)
achieve therapeutic aPTT in first 24hrs of therapy!
low molecular weight heparin (LMWH)
exoxaparin, dalteparin, tinzaparin, fondaparinux
binds AT–>inhibits 10a more than thrombin
renal excretion!
protamine reverses 60%
does not require aPTT monitoring
warfarin
inhibits vit K epoxide reductase–>inactivates factors 2, 7, 9, 10 protein C/S
liver metabolism
monitor with PT (factor 7 short half-life)
start within first 24hrs of treatment
anticoagulation therapy for acute coronary syndrome
unstable angina=Non-STEMI treatment
LMWH + aspirin (dec. risk of death, MI, and revascularization surgery)
anticoagulation therapy to prevent DVT/PE after orthopedic surgery
warfarin:
*
