Genetics and Syndromes Flashcards
Ewing sarcoma/PNET
5 translocations, all involving EWS gene at 22q12
- t(11;22)(q24;q12) Most common (>80%)
- EWS/Fli-1
- t(21;22)(q22;q12) Most of the remaining
- t(7;22)(p22;q12) Rare
- t(17;22)(q12;q12) Rare
- t(2;22)(q33;q12) Rare
Neuroblastoma
Neuroblastoma
- MYCN oncogene amplification
- PCR or FISH
- MYCN amplified tumors worse prognosis
Mesenchymal chondrosarcoma
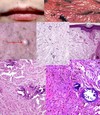
Mesenchymal chondrosarcoma
- sheets and clusters of uniform, small round cells
- multiple foci of well-differentiated cartilage
- t(11;33) (q24;q12) translocation
- expression of Sox9
- master regulator of cartilage differentiation
Immunohistochemistry to Detect Transforming Mutations?
Immunohistochemistry to Detect Transforming Mutations
- The point mutation from arginine to histidine in codon 132 (R132H) of isocitrate dehydrogenase 1 (IDH1) is commonly found in astrogliomas and is not present in gliosis. The test can be performed with an antibody specific to the mutant IDH1.
- Mutations of TP53 commonly lead to a protein with a longer half-life than the short-lived wild type p53 protein; thus accumulation of this protein can be studied by IHC.
- Integrase interactor 1 (INI-1) is deleted in rhabdoid tumors and epitheloid sarcomas; thus absence of the protein aids making the diagnosis of these entities.
- HER-2/NEU amplification is studied by Herceptest assessed according to ASCO-CAP or treatment of gastric cancer (ToG) guidelines for breast and gastric carcinoma, respectively
Thyroid Mutations
PTC
- BRAF (BRAFV600E)
- RAS genes (HRAS and NRAS)
- RET/PTC translocations (RET/PTC1 and RET/PTC3)
- TRK translocations (
Follicular thyroid carcinomas
- RAS (HRAS and NRAS codon61)
- PAX8/PPAR gamma translocations
Follicular adenomas
- RAS mutations
Medullary thyroid carcinoma
- Activating RET mutations
- Germ line mutations are seen in >95% of familial cases (MEN2 or familial medullary thyroid carcinoma).
Thyroid Mutations
BRAF
- PTC
RAS
- PTC
- Follicular carcinoma
- Follicular adenoma
RET
- PTC
- Medullary (MEN2 & familial medullary thyroid carcinoma)
TRK translocations
- PTC
PAX8/PPAR gamma translocations
- Follicular thyroid carcinomas
Mutations in PTC
Most common -> least common?
Pathway?
Associations?
Mutations in PTC
Activate the mitogen-activated protein kinase (MAPK) pathway
- BRAF point mutations
- 40-50%
- mainly BRAFV600E
- a/w classical and tall cell
- predict more aggressive behavior even in pT1 tumors
- only in 10% of follicular variant PTC
- RAS genes
- 10% to 20%
- mainly HRAS and NRAS codon 61 mutations
- mainly in the follicular variant
- RET/PTC translocations
- 10% to 20%
- fusions of RET with 11 different partners
- mainly RET/PTC1 [fusion partner CCDC6] and RET/PTC3 [fusion partner NCOA4]
- a/w classical histologic appearance, a younger age at diagnosis, lymph node metastasis, and radiation exposure (RET/PTC2)
- TRK translocations
*
Follicular Thyroid Mutations
Follicular Thyroid Mutions
RAS mutations
- up to 50% follicual carcinoma
- HRAS and NRAS codon61
- +/- follicular adenomas (precuresor?)
PAX8/PPAR gamma translocations
- 30-35% follicular carcinoma
Medullary Thyroid Carcinoma Mutations
Medullary Thyroid Carcinoma Mutations
- Activating RET mutations
- Germ line mutations un > 95% of familial cases
- MEN2
- familial medullary thyroid carcinoma
Thyroid FNA
Testing of thyroid fine needle aspirates
- BRAF V600E mutations, NRAS and HRAS codon 61 mutations, and RET/PTC translocations help manage thyroid nodules
- BRAF V600E mutation or RET/PTC translocationhas a high PPV for malignancy
- provided the LOD of the test is not
- BRAF V600E mutation or RET/PTC translocationhas a high PPV for malignancy
- PAX8-PPAR gamma translocation is strongly a/w invasion in a follicular neoplasm
- In specimens with indeterminate cytology, having 1 mutations is a/w an increased risk of malignancy
- 88% follicular lesion of uncertain significance
- 87% follicular neoplasm
- 95% suspicious for malignancy
- versus no mutation: 6%, 14%, and 28%
- the high PPV can allow total thyroidectomy instead of lobectomy in positive cases
Translocations in MALT lymphoma
Translocations in MALT lymphoma
t(11;18)
- API2 and MALT1 genes
- gastric and pulmonary
t(14;18)
- IgH and MALT1 genes
- ocular adnexa/orbit and salivary gland
Trisomy 3
- nonspecific abnormality frequently detected in MALT lymphomas
t(3;14)
- IgH and FOXP1 genes
- thyroid, ocular adnexa/orbit, and skin
Trisomy 18
- nonspecific abnormality frequently detected in MALT lymphomas
Associated mutation?

Trisomy 13
- least common of the viable autosomal trisomy syndromes (the others being Trisomy 18 and 21)
- phenotype typically includes cleft palate, polydactyly, microcephaly, and anomalies of the heart and kidneys
Adenoid cystic carcinoma (ACC) of the breast
- Incidence?
- Age?
- Hormone receptor profile?
- Clinical course?
- More aggressive behavior can be seen in the __ variant
- Morphologically, ACCs of the breast are similar to those seen in salivary glands, composed of two cell types
- one which stains with luminal epithelial markers ____ (4)
- the other staining with ___ (4) and ___(4)
- ACCs of the salivary gland and breast consistently harbor the hallmark __ fusion gene resulting from a __ translocation.

Adenoid cystic carcinoma (ACC) of the breast
- rare tumor accounting for less than 0.1% of all breast carcinomas
- occurs predominantly in postmenopausal women (mean age of 60)
- triple negative (ER/PR/HER2) receptor profile
- indolent clinical course, presenting with localized disease
- more aggressive behavior can be seen in the solid variant
- Morphologically, ACCs of the breast are similar to those seen in salivary glands, composed of two cell types
- one which stains with luminal epithelial markers CK7, EMA, CEA, and CD117
- the other staining with high molecular weight/basal cytokeratins CK5, CK5/6, CK14, CK17 and myoepithelial markers p63, S100, actin, calponin
- ACCs of the salivary gland and breast consistently harbor the hallmark MYB–NFIB fusion gene resulting from a t(6;9)(q22–23;p23–24) translocation.
- Prognosis?
- Location?
- Age?
- Clinical presentation?
- Frequency of recurrence?
- Cell of origin?
- Genetics?
- Most helpful diagnostic marker? caveat?
- Best way to detect mutation?
DDx:

Dermatofibrosarcoma Protuberans
- slow-growing dermal spindle cell tumor of intermediate malignancy
- typically occurs on the trunk and proximal extremities of young and middle-aged adults
- solitary lesion or multiple polypoid nodules arising in an indurated plaque
- Local recurrence is common, occurring in one third of all cases.
- immunohistochemical and ultrastructural evidence indicates a fibroblastic origin
- t(17;22) translocation in > 90% –> pathogenic COL1A1-PDGFB fusion gene
- most helpful diagnostic marker is CD34, which stains 50% to 100% of the cells
- CD34 is negative or focally positive in dermatofibromas
- PCR may be used to identify the COL1A1-PDGFB fusion gene
DDx:
Giant cell fibroblastoma
- considered a variant of dermatofibrosarcoma protuberans that often manifests in childhood
- may exhibit the same immunogenetic and cytogenetic profile as DFSP
Fibrosarcomatous change in DFSP
- characterized by a fascicular or herringbone growth pattern, represents malignant transformation
- fibrosarcomatous areas express CD34 in less than 50% of cases
Stains?
EM?
Mutation?
Acute disseminated form?
2 other types?

Langerhans Cell Histiocytosis
- Langerhans cell histiocytosis previously was divided into multiple clinical subtypes, but there is much overlap. The different forms of Langerhans cell histiocytosis have similar pathology in that there is a proliferation of Langerhans cells, which can be identified by the distinctive reniform, or kidney-shaped, nuclei.
- Langerhans cells stain positively with S-100 protein and CD1a immunohistochemical stains. Langerhans cells do not stain with CD68.
- With electron microscopy, characteristic Birbeck granules are seen within Langerhans cells. The Birbeck granules resemble a tennis racquet.
- BRAF V600Emutation
Letterer-Siwe disease
- acute disseminated form of Langerhans cell histiocytosis
- usually seen in infants and has many systemic manifestations
- skin lesions are characterized by hyperpigmented scaly patches, which can coalesce and form a seborrheic dermatitis–like eruption
- unresponsive dermatitis in the diaper area
- hyperpigmented papules with associated hemorrhage at the periphery of the dermatitis area
Hand-Schüller-Christian disease
- a form of Langerhans cell histiocytosis that is characterized by the triad of bone lesions, exophthalmos, and diabetes insipidus
Eosinophilic granuloma
- a form of Langerhans cell histiocytosis that usually consists of either a few lesions or one lesion and most commonly affects the bone
- skin and oral mucosa occasionally can be involved.
Congenital self-healing reticulohistiocytosis
- also known as Hashimoto-Pritzker disease
- a variant of Langerhans cell histiocytosis with a very good prognosis
- lesions are often present at birth but can manifest within the first few weeks of life as well
- most commonly, there are scattered papules and nodules over the skin
- occasionally, only a single lesion is present
Located in lateral ventricle
a/w?
grade?
histology?

Subependymal giant cell astrocytoma (SEGA)
- Associated with Tuberous Sclerosis
- most common CNS neoplasm in TS patients
- typically present in the wall of the lateral ventricles and are predominantly exophytic
- benign, slow-growing, discrete neoplasms and are graded as WHO grade I tumors
- many large plump cells with abundant glassy eosinophilic cytoplasm and large eccentric nuclei are present accompanied by elongated cells with smaller nuclei and fibrillar processes, forming a nodular appearance
Tuberous sclerosis
CNS lesions?
Gross?
Histology?
Most common?
Genes?

Tuberous sclerosis lesions in CNS
- cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas
- patients can present with seizures, mental retardation, behavioral problems and raised intracranial pressure
Cortical tubers
- effacement of the neocortex with collections of large, bizarre cells and extensive fibrillary gliosis beneath the pia
- Grossly, the cortical tubers are firm and scattered over the brain blurring the gray-white matter junction
Subependymal nodules
- firm or calcified protrusions, single or in rows, more commonly in the walls of the lateral ventricles
Subependymal giant cell astrocytomas (SEGA)
- most common CNS neoplasm in TS patients
- typically present in the wall of the lateral ventricles and are predominantly exophytic
- benign, slow-growing, discrete neoplasms
- WHO grade I tumors
- many large plump cells with abundant glassy eosinophilic cytoplasm and large eccentric nuclei are present accompanied by elongated cells with smaller nuclei and fibrillar processes, forming a nodular appearance
TSC1 gene: hamartin, 9q34
TSC2 gene: tuberin, 16p13.3

Tuberous sclerosis cutaneous lesions

Tuberous sclerosis cutaneous lesions
- hypomelanic macules (ash-leaf)
- facial angiofibromas
- periungal or subungal fibromas
- shagreen patches
- fibrous hamartomas
Two different genes are associated with tuberous sclerosis:
- TSC1 gene: hamartin, 9q34
- TSC2 gene: tuberin, 16p13.3

Tuberous sclerosis
Inheritance pattern?
Lesions?
Presentation?
Genes?
Tuberous sclerosis
- autosomal dominant
- lesions in the CNS
- cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas
- patients can present with seizures, mental retardation, behavioral problems and raised intracranial pressure
- cutaneous lesions
- hypomelanic macules, facial angiofibromas, periungal or subungal fibromas, and shagreen patches or fibrous hamartomas
- cardiac rhabdomyomas
- pulmonary lymphagioleiomyomatosis
- renal angiomyolipomas, and renal cysts
- Two different genes are associated with tuberous sclerosis:
- TSC1 gene: hamartin located in chromosome 9q34
- TSC2 gene: tuberin located in chromosome 16p13.3
Age?
Gender?
Location?
Presentation (early and late)?
Prognosis?
Tx?
What reduces recurrence?
Gross?
Histology?
Stain?
What counts as fibrosarcomatous transformation?
Most common site of dissemination?
Mutation?

Dermatofibrosarcoma protuberans
- young adults
- male predominance
- trunk and the proximal extremities
- early lesions have a plaquelike appearance and late lesions are multinodular with skin ulceration
- low-grade sarcoma with a high propensity for local recurrence if incompletely excised
- complete excision requires wide margins because of infiltration beyond the grossly visible margins
- recurrence rate of 20% within 2 years of surgery
- Mohs surgery significantly reduces the rate of recurrence (
- Gross: the lesion is firm and fibrous and varies from a small dermis based plaquelike area or nodule to a large multinodular lesion that ulcerates the overlying skin and deeply involves the underlying adipose tissue
- occasionally may be purely subcutaneous
- Histology:
- proliferation of uniform, mildly atypical spindle cells, arranged in a tight, repetitive storiform pattern
- infiltrates the dermis surrounding the epidermal appendages and infiltrates the fat in a checkerboard or beaded pattern
- lesional cells are uniformly CD34 positive
- may contain fascicular areas that are indistinguishable from fibrosarcoma
- composed of intersecting fascicles of spindle cells with increased atypia and mitotic activity (>10 mitoses per 10 HPFs)
- to be considered fibrosarcomatous, must represent > 5% of the entire lesion
- DFSP with fibrosarcomatous transformation has a small but definite metastatic potential 5% to 10%
- lung is the most common site of dissemination
- t(17;22) COL1A1-PDGFB
__of chromosome __ is seen in 80% of T-cell prolymphocytic leukemia cases.
Inversion of chromosome 14 is seen in 80% of T-cell prolymphocytic leukemia cases.
50 yo female with scalp mass.

Epithelioid Hemangioendothelioma
- myxoid background
- epithelioid cells in chains or nests
- blister cells trying to make vascular channels but can’t
- translocation WWTR1-CAMTA1, YAP1-TFE3 fusion gene




Clinical?
Histology?
Genetics?
Tx?

DARIER’S DISEASE
Mige the dominatrix has an ATP2A2 (aptitude) for shoving greasy corps ronds and grains up Darier’s
- Clin: greasy papules on head and neck
- Acanthosis, suprabasal acantholytic dyskeratosis with “corps ronds” (dyskeratotic cells) and grains (parakeratotic cells)
- Genetics:
- AD
-
ATP2A2, 12q23-24.1, encodes the sarcoplasmic/endoplasmic reticulum Ca2+ -ATP isoform 2 protein (SERCA2)
- This pump maintains a low cytoplasmic Ca2+ level by actively transporting calcium ions from the cytosol into the lumen of the endoplasmic reticulum.
- Miglustat: orphan drug, α-glucosidase inhibitor, restores mature adherens junctions and desmosomes and increases adhesion strength
Clinical?
Histology?
Genetics?
Tests?

HAILEY-HAILEY DISEASE (BENIGN FAMILIAL PEMPHIGUS)
Hailey’s comet hit a dilapedated brick wall, ATP2C1 (a teepee and 2 C1’s) but you couldn’t tell the DIF-ference
- Vesicles and erythematous plaques with overlying crusts typically occur in the genital area, as well as the chest, neck, and axillary
- “dilapidated brick wall” with full-thickness and suprabasilar acantholysis
- Defect in a calcium pump protein, ATP2C1 which encodes the secretory pathway Ca2+/Mn2 ATPase (hSPCA1).
- loss of sensitivity to Ca2+ and Mn2+ion binding and transport
- low levels of Ca2+ within Golgi bodies impair protein processing
- DIF is negative

Thin Basement Membrane Nephropathy
- The clinical presentation of microscopic hematuria, in the absence of significant proteinuria or renal insufficiency, is most commonly encountered in the following three renal conditions: thin basement membrane nephropathy (TBMN), hereditary nephritis (i.e., Alport syndrome), and IgA nephropathy.
- Before a patient with microscopic hematuria undergoes renal biopsy, possible urologic causes for hematuria must be excluded.
- The glomeruli in TBMN usually appear unremarkable on light microscopy. The normal glomerular basement membrane (GBM) thickness averages approximately 320 nm in women and 350 nm in men. Diagnostic criteria for TBMN are diffuse GBM thinning, with mean thickness less than 225 nm in women and less than 250 nm in men.
- The main clinical finding in TBMN is isolated microscopic hematuria. Many patients have a family history of isolated hematuria with autosomal dominant transmission.
- TBMN results from a heterozygous mutation in either COL4A3 or COL4A4; these genes encode the alpha-3 (COL4A3) and alpha-4 (COL4A4) chains of collagen IV.
- TBMN is a nonprogressive condition that is often referred to clinically as benign essential hematuria.
Dx?
Histology?
Subtypes?
Stains?
Children and young adults with multiple__ and diabetes?
Most likely to progress an special staining?

Hepatocellular Adenoma (Liver Cell Adenoma)
- benign tumor of hepatocytes
- Histology
- thickened cords of hepatocytes
- interspersed blood vessels (thin venous and arterial profiles are seen)
- NO bile ducts
- subclassified into four groups based on genomic classification:
- type 1, HNF-1α mutations, with large droplet steatosis and without atypia
- type 2, β-catenin mutations, with atypia and features that may transition to hepatocellular carcinoma
- type 3, interleukin-6 ST mutations, including inflammatory or telangiectatic adenoma
- type 4, no known mutation, with no specific features
- to distinguish it from carcinoma
- reticulin (which should show preserved reticulin framework throughout, in contrast to the “paucireticulin” pattern seen in hepatocellular carcinoma
- possibly CD34 (should show limited expression near pseudo–portal tracts).
- immunostains can be added
- trio of glypican-3, glutamine synthetase, and heat shock protein 70 (which all should be negative)
- Children and young adults with multiple adenomas (adenomatosis is defined as ≥10 adenomas) and diabetes are very likely to haveHNF-1α mutation and maturity-onset diabetes of the young type 3.
- Adenomas with suspicious features for the development of or transition to hepatocellular carcinoma (nuclear atypia, pseudoglandular growth, increased mitotic activity, focal loss of reticulin) and harboring β-catenin mutations should demonstrate glutamine synthetase expression and nuclear overexpression of β-catenin on immunohistochemical staining.
Neurodegenerative diseases, genetic abnormalities
• A large number of neurodegenerative diseases can be caused by genetic mutations or abnormalities. Genetic abnormalities can include single gene mutations and microsatellite repeat instability leading to expansion of tandem triplet repeats.
Mutations in the gene for α-synuclein, SNCA, cause __
In Huntington disease, an increase in the number of repeats of the __ sequence is seen in the__ gene localized in the __ and coding for the protein __.
Mutations in the __ gene have been associated with autosomal dominant forms of Alzheimer disease. The __ gene is located on chromosome 21 (thus, the association of Alzheimer disease and Down syndrome) and codes for the __ protein.
- The tau gene, __, is located on chromosome__ and codes for the __ protein. Mutations in this gene are associated with__.
- In X-linked bulbospinal neuronopathy (like Kennedy disease), a __ causes slowly progressive lower motor neuron weakness of facial, bulbar, and proximal limb muscles.
Neurodegenerative diseases, genetic abnormalities
- A large number of neurodegenerative diseases can be caused by genetic mutations or abnormalities. Genetic abnormalities can include single gene mutations and microsatellite repeat instability leading to expansion of tandem triplet repeats.
- Mutations in the gene for α-synuclein, SNCA, cause some familial cases of Parkinson’s disease. α-synuclein is a protein that forms the pathologic inclusions (Lewy body) in Parkinson’s disease, diffuse Lewy bodies, and multiple system atrophy.
- In Huntington disease, an increase in the number of repeats of the CAG sequence is seen in the HTT gene localized in the short arm of chromosome 4 and coding for the protein huntingtin.
- Mutations in the APP gene have been associated with autosomal dominant forms of Alzheimer disease. The APP gene is located on chromosome 21 (thus, the association of Alzheimer disease and Down syndrome) and codes for the amyloid-β precursor protein (APP).
- The tau gene, MAPT, is located on chromosome 17 and codes for the microtubule-associated protein, tau. Mutations in this gene are associated with chromosome 17-linked frontotemporal lobar dementia.
- In X-linked bulbospinal neuronopathy (like Kennedy disease), a CAG repeat region in the first exon of the androgen receptor gene on the X chromosome causes slowly progressive lower motor neuron weakness of facial, bulbar, and proximal limb muscles.
Dx?
Age?
Gender?
Prognosis?
Staining?
Genetics?

Salivary duct carcinoma
- > 50 yo
- M > W
- most aggressive salivary gland malignancies
- perineural invasion and lymph node involvement occur in the majority
- occurs de novo and/or within preexisting pleomorphic adenoma (i.e., as a manifestation of carcinoma ex pleomorphic adenoma)
- positive nuclear immunohistochemical staining for androgen receptor, and positive membranous staining for Her-2/neu
- 17q21.1 amplification, involving the gene ERBB2, 40% of salivary duct carcinoma
Which disorder, also known as Duncan disease, is characterized by hepatic necrosis with a profound NK/T cell infiltrate?
X-LINKED LYMPHOPROLIFERATIVE DISORDER.
As an X-linked disorder, primarily men are affected. The range that disease can manifest extends from the previously mentioned hepatic necrosis and death to less severe agammaglobulinemia or B-cell lymphoma. The disorder is due to a defect in the SAP gene, which leads to uncontrolled NK/T cell activation.

SUPERFICIAL ANGIOMYXOMA
- Primarily young adults
- Trunk, lower extremities, head & neck
- Generally solitary, can be multiple in cases of Carney complex (endocrinopathy, mucocutaneous pigmentation, myxoma)
- Multilobulated growth, abundant stromal mucin, curvilinear vasculature, stromal,neutrophils
- Differential diagnosis: low-grade myxoid MFH
Carney complex
“CEMP” - Carney CEMPlex, young white people go to CEMP, the PRKA1 and the Carney-val with their families
-
Cutaneous melanocytic lesions
- Blue nevi, Lentigenes
-
Endocrine hyperplasia/neoplasia
- Pituitary
- Adrenal cortex
- Thyroid
- Testis: Large Cell Calcifying Sertoli Cell Tumor
- Myxomas: Cutaneous, Cardiac, External ear, Breast myxoid fibroadenoma
-
Psamomatous melanocytic schwannomaa
- GI, Cardiac, Peripheral nerves
- Genetics
- AD, PRKAR1-alpha
- 10-20 yo, white (any age/gender)
BRAF MUTATIONS
BRAF MUTATIONS
- PTC
- Melanoma
- Cholangiocarcinoma
- GBM
Activating mutation in exon 15
Valine > glutamate (V600E)

MEN Type 2
- MEN 2A > FMTC > MEN 2B
- Genetics
- AD
- RET proto-oncogene, 10q11.2
- MEN 2A
- Medullary thyroid carcinoma (less virulent)
- virtually all pts
- Pheochromocytoma
- 50% of pts, 60-80% bilateral
- Parathyroid hyperplasia
- < 50% of pts
- Medullary thyroid carcinoma (less virulent)
- MEN 2B
- Medullary thyroid carcinoma (more virulent)
- Pheochromocytoma
- Mucocutaneous ganglioneuromas
- Marfanoid habitus
- FMTC (familial medullary thyroid carcinoma) only
- Medullary thyroid carcinoma
GORLIN SYNDROME - NEVOID BCC SYNDROME

GORLIN SYNDROME - NEVOID BCC SYNDROME
- Genetics
- Autosomal dominant
- PTCH gene on chromsome 9q22.3 (regulateds Hedgehog pathway signaling)
- “2 hit” hypothesis
- Skin:
- multiple BCC before age 20
- palmar and plantar pits
- Nervous system:
- medulloblastoma
- Others:
- odontogenic keratocysts
- ovarian fibroma
- skeletal abnormalities

Familial Tumor Syndromes Involving the nervous System
Familial Tumor Syndromes Involving the nervous System
- Neurofibromatosis type I
- Neurofibromatosis type 2
- Li-Fraumeni Syndrome
- Turcot Syndrome
- Gorlin Syndrome (Naevoid Basal Cell Carcinoma Syndrome)
- Von Hippel-Lindau
- Tuberous Sclerosis
- Cowden Syndrome
- Gene:
- Chromosome:
- Neurvous system:
- Skin:
- Others:

Neurofibromatosis type I
- Gene: NF1
- Chromosome: 17q11
- Neurvous system:
- Neurofibroma
- MPNST
- optic nerve glioma
- astrocytoma
- PSAMMOMATOUS CARCINOID
• Almost exclusively seen in the
ampullary/periampullary location
- Skin:
- Café-au-lait spots
- axillary freckling
- Others:
- iris hamartomas
- osseous lesions
- phaeochromocytoma
- leukaemia
- Gene:
- Chromosome:
- Neurvous system:
- Others:

Neurofibromatosis type 2
- Gene: NF2
- Chromosome: 22q12
- Nervous system:
- schwannoma
- meningiomas
- meningioangiomatosis
- spinal ependymoma
- astrocytoma
- Others:
- posterior lens opacities
- retinal hamartoma
- Gene:
- Chromosome:
- Nervous system:
- Others:

Li-Fraumeni Syndrome
- Gene: TP53
- Chromosome: 17p13
- Nervous system:
- astrocytomas
- PNET
- Others:
- breast ca
- bone and soft tissue sarcoma
- adrenocortical ca
- leukaemia
- Gene:
- Chromosome:
- Nervous system:
- Others:

Von Hippel-Lindau
- Gene: VHL
- Chromosome: 3p25
- Nervous systme:
- hemangioblastoma
- Others:
- retinal hemangiolastoma
- renal cell carcinoma
- pheochromocytoma
- visceral cysts
- Immunostain: inhibin and CD10
- Gene:
- Chromosome:
- Nervous system:
- Others:

Turcot Syndrome
- Gene: APC
- Chromosome: 5q21
- Nervous system:
- medulloblastoma
- Others:
- colorectal polyps
- Gene: hMLH1
- Chromosome: 3p21
- Nervous system:
- glioblastoma
- Others:
- colorectal polyps
- hPSM
Gene:
Chromosome:
Nervous system:
Skin:
Others:

Tuberous Sclerosis
- Gene: TSC1 and TSC2
- Chromosomes: 9p34 and 16p13
- Nervous system:
- subependymal giant cell astrocytoma
- subependymal nodules
- cortical tubers
- Skin:
- cutaneous angiofibroma
- subungual fibroma
- fibrous hamartoma
- hypopigmented macule
- shagreen patch
- Others:
- cardiac rhabdomyoma
- adenomatous polyps of the duodenum and the small intestine
- cysts of the lung and kidney
- lymphangioleiomyomatosis
- renal angiomyolipoma
Gene:
Chromosome:
Nervous system:
Skin:
Others:

Tuberous Sclerosis
- Gene: TSC1 and TSC2
- Chromosomes: 9p34 and 16p13
- Nervous system:
- subependymal giant cell astrocytoma
- subependymal nodules
- cortical tubers
- Skin:
- cutaneous angiofibroma
- subungual fibroma
- fibrous hamartoma
- hypopigmented macule
- shagreen patch
- Others:
- cardiac rhabdomyoma
- adenomatous polyps of the duodenum and the small intestine
- cysts of the lung and kidney
- lymphangioleiomyomatosis
- renal angiomyolipoma

Gene:
Chromosome:
Nervous system:
Skin:
Others:

Tuberous Sclerosis
- Gene: TSC1 and TSC2
- Chromosomes: 9p34 and 16p13
- Nervous system:
- subependymal giant cell astrocytoma
- subependymal nodules
- cortical tubers
- Skin:
- cutaneous angiofibroma
- subungual fibroma
- fibrous hamartoma
- hypopigmented macule
- shagreen patch
- Others:
- cardiac rhabdomyoma
- adenomatous polyps of the duodenum and the small intestine
- cysts of the lung and kidney
- lymphangioleiomyomatosis
- renal angiomyolipoma

Gene:
Chromosome:
Nervous system:
Skin:
Others:

Cowden Syndrome
- Gene: PTEN
- Chromosomes: 10q23
- Nervous system:
- dysplastic gangliocytoma of the cerebellum (Lhermitte-Duclos)
- megalencephaly
- Skin:
- multiple trichilemmoma
- fibroma
- Others:
- hamartomatous polyps of the colon
- thyroid neoplasms
- endometrial cancers
- breast carcinoma

Carney Triad/Syndrome
“PEG” - Young orphan female Carneys like PEGging and sucking D (Big)
- Pulmonary Chondroma
- Extra-adrenal Paraganglioma
- GIST (gastric, epithelioid)
- Genetics
- Succinate dehydrogenase deficiency (SDHB)
- Few have CKIT or PDGFRA
- Not Familial
- < 30 yo
- F > M

Carney-Stratakis Syndrome
- Familial Paraganglioma and GIST
- Paraganglioma - aggresive
- GIST - indolent, gastric
- Genetics
- Germline mutation in succinate dehydrogenase gene
- SDHB, SDHC, SDHD
- 23 yo mean age
- M > F
MEN-1

MEN-1 (Multiple Endocrine Neoplasias Type 1)
aka Wermer syndrome (Pituitary, Parathyroid, Pancreatic islet cell)
- Pituitary tumors (anterior)
- Secrete prolactin (60%), growth hormone (25%), corticotropin (< 5%), the rest nonfunctional
- Parathyroid tumors
- 90% primary hyperparathyroid (most common presentation)
- hypercalcemia, nephrolithiasis, and bone abnormalities (osteitis fibrosa cystica)
- multiple parathyroid glands, younger, high recurrence vs. non-MEN1
- Pancreatic Islet Cell tumors
- 30-80% (2nd most common)
- Nonfunctional is most common
- Gastrinomas > insulinomas, glucagonomas, vasoactive intestinal polypeptidomas (VIPomas), and pancreatic polypeptidomas (PPomas)
- often multicentric +/- malignant transformation
- Other:
- Carcinoid, subcutaneous lipoma, facial angiofibromas, collagenomas, adrenal tumors, thyroid adenomas
- Genetics
- MEN gene, 11q13 loss of heterozygosity (tumor suppressor gene)
- inherit one mutated copy and require a somatic mutation of the second copy for tumor development
- AD or sporadic
- 50% penetrant by 20 yo, 95% by 40 yo
- M=F, presents 20’s in F, 30’s in M
- 50% mortality by age 50
*

