Chapter 5: Genetic Disorders Flashcards
Disorders related to mutations in single genes with large effects are also called?
What is their pentrance and how common?
- Mendelian disorders
- Uncommon, but highly penetrant
What plays an important role in the pathogenesis of complex multigenic disorders/multifactoral disorders?
- Enviornmental factors
- Each polymorphism has a small effect and is of low penetrance, but the more that exist the higher the risk becomes
Atherolsclerosis, diabetes, HTN, autoimmune diseases, and even normal traits such as height and weight are governed by?
Polymorphisms in several genes
The sickle mutation affecting the β-globin chain of hemoglobin is an example of what type of mutation?
What is the change in the AA sequence?
- Non-conservative missense mutation
- CTC (or GAG in mRNA) = glutamic acid —-> CAC (GUG in mRNA) = valine
β0-thalassemia, a rare form of anemia, is due to what kind of point mutation?
What is the change in AA sequence?
- Nonsense mutation (stop codon)
- CAG (glutamine) —> UAG; creates stop codon
- Premature termination of β-globin gene translation = short peptide that is rapidly degraded
What is the distinguishing feature of trinucleotide-repeat mutations?
They are dynamic (i.e., the degree of amplification increases during gametogenesis)
A 4 base insertion in the hemosaminidase A gene, leads to what type of mutation and is the major cause of what disease?
- Frameshift mutation
- Tay-Sachs disease in Ashkenazi Jews
What type of mutation is responsible for the ABO type O?
- Frame-shift mutation
- Single base deletion at the ABO (glycosyltransferase) locus
What differentiates sick cell disease from sickle cell trait?
- Sickle cell disease: homozygote – all the hemoglobin is HbS
- Sickle cell trait: heterozygot – some hemoglobin is HbS and rest is normal HbA – red cell sickling only occurs under circumstances such as exposure to lowered oxygen tension
How is sickle cell anemia an example of pleiotropism?
- Point mutation in gene gives rise to HbS, predisposing red cells to hemolysis, which tend to cause a logjam in small vessels
- Can lead to splenic fibrosis, organ infarcts, and bone changes
- Numerous differing end-organ derangements are all related to the primary defect of Hb synthesis
How can someone have an autosomal dominant disorder, without having at least one affected parent?
Proportion of patients who develop the disease as a result of a new mutation is related to?
Who is more likely to be the contributor of a disease due to a new mutation?
- Mutations involving either the egg or the sperm from which they were derived
- Depends on the effect of the disease on reproductive capability. If disease markedly reduces repro. fitness, most cases would be expected to result from new mutations
- Many new mutations seem to occur in germ cell of older fathers.
A trait seen in all individuals carrying a gene but is expressed differently among individuals is known as?
What controls this variability?
- Variable expressitivity
- Effects of other genes or the enviornment modify the phenotypic expression
Many autosomal dominant disease arising from deleterious mutations affecting what 2 types of biochemical mechanisms/proteins?
- Those involved in regulation of complex metabolic pathways that are subject to feedback inhibition (i.e., membrane receptors – LDL receptor)
- Key structural proteins, such as collagen and cytoskeletal elements of the red cell membrane (i.e., spectrin)
Why does even a single mutant collagen chain have such a large effect?
What is the mutant allele in this case known as?
- Collagen molecules are trimers and require 3 collagen chains arranged in a helical configuration
- Each chain in the helix MUST be normal for the assembly and stability of the collagen molecule
- Known as a dominant negative because it impairs the function of a normal allele
Gain of function mutations are almost always what type of inheritance pattern?
Which disease illustrates this type of mutation?
- Autosomal dominant
- Huntington’s disease gives rise to abnormal protein, huntingtin, that is toxic to neurons, and hence even heterozygotes develop neurologic deficit
What are the 4 diseases that are autosomal dominant and affect the nervous system? (hint: there is a mnemonic)
- Tuberous sclerosis
- Myotonic dystrophy
- Huntington disease
- Neurofibromatosis
Touch My Hurt Nerves
Autosomal dominant disorders affecting the Urinary and GI systems?
Urinary = Polycystic kidney disease
GI = familial polyposis coli
What are the 4 diseases affecting the skeletal system that are Autosomal Dominant? (hint: there’s a mnemonic)
1) Marfan Syndrome
2) Osteogenesis imperfecta
3) Ehlers-Danlos syndrome (some variants)
4) Achondroplasia
“My Osteology Enters Afterlife”
What are the 2 diseases affecting the metabolic system that are Autosomal Dominant?
- Familial hypercholesterolemia
- Acute intermittent porphyria
What kind of penetrance is common with autosomal recessive disorders?
How are parents and children affected by these disorders?
- Complete penetrance
- Trait does not usually affect the parents (carriers), siblings have 1/4 chance of having trait.
Many of the mutated genes in autosomal recessive disorders affect which proteins?
Enzymes
Almost all inborn errors of metabolism follow what type of inheritance?
Autosomal Recessive
If a male is affected by an X-linked disorder, they are said to be ________ for X-linked mutant genes
Hemizygous
How are X-linked recessive disorders passed down from an affected male?
Carrier mother?
- A male will pass on to all his daughters, and they will be carriers. Will not pass to his sons.
- A heterozygous mother will pass to 50% of her sons and/or daughters
How are X-linked dominant conditions passed to offspring from both males and females?
- Affected heterozygous female will pass to half her sons and half her daughters
- Affected male will pass to all his daughters, but none of his sons, if female parent unaffected
Which condition shows that although a mutant X chromosome may be inactive in some cells, it may be active in other cells?
Affect of drugs?
Who’s at greatest risk (male or female)?
- Glucose 6-phosphate dehydrogenase (G6PD) deficiency
- Predisposes patients to RBC hemolysis when they are treated with certain drugs (i.e., primaquine, anti-malarial) = severe drug-induced hemolytic reaction
- Males more affected because they only have one X chromosome and if gene is mutant they have no functional G6PD. Females have two X chromosomes.
X-linked recessive diseas that affects the MSK system?
Duchenne muscular dystrophy
X-linked recessive disorders that affect the blood system (3 of them)?
1) Hemophilia A and B
2) Chronic granulomatous disease
3) G6PD deficiency
What 2 X-linked recessive disorders affect the immune system?
1) Agammaglobinemia
2) Wiskott-Aldrich syndrome
What 2 X-linked recessive disorders affect the metabolic system?
1) Diabetes insipidus
2) Lesch-Nyhan syndrome
What X-linked recessive disorder affects the nervous system?
Fragile X syndrome
What is the mode of inheritance for Vitamin D-resistant rickets?
X-linked dominant
Galactosemia is due to a deficiency in ________, leading to the accumulation of galactose and consequent tissue damage?
Galactose-1-phosphate uridyltransferase
α1-antitrypsin deficiency leads to what?
- Inability to inactivate neutrophil elastase in the lungs
- Leads to destruction of elastin in the walls of lung alveoli, and eventually pulmonary emphysema
What are the 2 fundamental mechanisms by which loss of fibrillin leads to clinical manifestations of Marfan syndrome?
1) Loss of structural support in microfibril rich CT
2) Excessive activation of TGF-β signalling
Fibrillin occurs in what two homologous forms?
Mapped to which chromosomes?
- Fibrillin-1 (FBN1) mapped to chromosome 15q21.1
- Fibrillin-2 (FBN2) mapped to chromosome 5q23.31
What type of mutations give rise to the abnormal fibrillin-1 seen in Marfan syndrome?
Missense mutations
Mutations of FBN2 are less common and give rise to?
Congenital contractural arachnodactyly
How does loss of microfibrils give rise to abnormal and excessive activation of TGF-β?
What does this excessive activation lead to?
- Normal microfibrils sequester and control the bioavailability of this cytokine
- Deleterious effects on vascular smooth muscle development and increases activity of MMPs, causing loss of ECM
Finding of bilateral ectopia lentis should raise suspicion of which disease?
What is ectopia lentis?
- Marfan Syndrome; since is so uncommon in persons w/o this disease its presence is nearly diagnostic
- Bilateral subluxation or dislocation (outward and upward) of the lens
Most cases of Marfan Syndrome transmitted via what type of inheritance?
Autosomal dominant
What are the most life threatening lesions seen in Marfan Syndrome; what are the 2 most common?
How can they be detected?
- Cardiovascular lesions; 2 most common are mitral valve prolapse and more importantly dilation of the ascending aorta due to cystic medionecrosis
- Echocardiography is extremely valuable in diagnosis
Due to variations, the clinical diagnosis of Marfan syndome is currently based on?
What are the guidelines for using these criteria?
- Revised Ghent criteria
- Takes into account family hx, cardinal clinical signs in absence of family hx, and presence of fibrillin mutation
- In general, major involvement of 2 of the 4 organ systems (skeletal, cardiovascular, ocular, and skin) and minor involvement of another organ is required for diagnosis
What is the mainstay of the medical treatment for Marfan syndrome?
Other treatments being tested?
- Mainstay = β blockers; act to reduce heart rate and aortic wall stress
- Other therapies being tested = block TGF-β signaling and blockade of angiotensin type 2 receptors
Heterogenous group of conditions that result from a defect in the synthesis of fibrillar collagen?
Ehlers-Danlos Syndrome
What are the 2 autosomal recessive types of Ehlers-Danlos syndrome?
What is the gene defect in each?
1) Kyphoscoliosis (Type VI) due to Lysyl hydroxylase defect = most common autosomal recessive form
2) Dermatosparaxis (Type VIIc) due to Procollagen N-peptidase defect
Vascular type (IV) of EDS arises from abnormalities in?
Which gene
- Type III collage
- COL3A1 gene
Which tissues are rich in type III collagen and are affected most by vascular type of EDS?
Blood vessels and intestines
The arthrochalasia type and dermatosparaxis type of EDS arise from defects in?
Which genes is defective for each type
- Conversion of type I procollagen to collagen; through cleavage of noncollagen peptides from the N and C terminus of the procollagen
- Arthrochalasia type = mutations in either COL1A1 or COL1A2
- Dermatosparaxis type = mutation in procollagen-N-peptidase gene
The classic type of EDS arises from mutations in what genes?
Can also be caused by non-collagen related gene abnormalities such as?
Mutations in tenascin-X lead to?
- Genes for type V collagen (COL5A1 and COL5A2)
- Defects that affect the biosynthesis of other extracellular matrix molecules that influence collagen synthesis
- EDS-like condition caused by mutation in tenascin-X, a large multimeric protein, that affects synthesis and fibril formation of type VI and type I collagens
How does familial hypercholesterolemia differ amongst homozygotes and heterozygotes?
Heterozygotes: have one mutant gene, with 2-3x elevation of blood cholesterol levels, leads to tendinous xanthomas and premature atherosclerosis
Homozygotes: have 2 mutant genes and 5-6x elevation of blood cholesterol levels. May develop skin xanthomas and coronary, cerebral, and peripheral vascular atherosclerosis at a early age
VLDLs released by liver are rich in ______ and contain lesser amounts of _______
VLDLs released by liver are rich in triglycerides and contain lesser amounts of cholesterol esters
Which receptor on the liver recognizes IDL and specifically what does it recognize?
- LDL-receptor
- Recognizes apo B-100 and apo-E
What is the immediate and major source of plasma LDL?
IDL
Which apopprotein is found on LDL and can be recognized by the LDL-recptor for uptake/clearance by the liver?
ApoB-100
The exit of cholesterol from the lysosomes requires the action of what 2 proteins?
NPC1 and NPC2
Cholesterol suppresses cholesterol synthesis within the cell by inhibiting?
Also suppresses the synthesis of?
- HMG-CoA reductase = The rate-limiting enzyme
- Suppresses synthesis of LDL receptors, thus protecting cells from excessive accumulation of cholesterol
Statins work by suppressing what?
But increasing?
- Suppress intracellular cholesterol synthesis by inhibiting enzyme HMG-CoA reductase
- Allows greater synthesis of LDL receptors
LDL can also be transported via scavenger receptors, which occurs via what cells?
How does this contribute to the pathogenesis of hypercholesterolemia?
- Cells of the mononuclear phagocyte system; monocytes and macrophages have receptors for chemically altered (i.e., acetylated or oxidized) LDL.
- Impaired IDL transport into liver secondarily diverts more plasma IDL into precursor pool for LDL
- There is marked increased in the scavenger receptor-mediated traffic of LDL cholesterol into the cells of this system and possibly the vascular walls = appearance of xanthomas and premature atherosclerosis
Differentiate class I vs. class II mutations of the LDL receptor gene?
Class I: uncommon; complete failure of synthesis of the receptor protein
Class II: fairly common; encode receptor proteins that accumulate in the ER because folding defects make it impossible for them to be transported to the Golgi
Differentiate class III vs. class IV vs. class V mutations of the LDL receptor gene?
Class III: affect LDL-binding domain of receptor; encoded protein reaches cell surface, but fails to bind LDL
Class IV: bind LDL normally, but fail to localize in coated pits and bound LDL is not internalized
Class V: bind LDL and can be internalized; however pH-dependent dissociation of receptor and bound LDL fails to occur, trapped in endosome, and fail to recycle
What are 3 treatment strategies for lysosomal storage disease?
- Enzyme replacement therapy - currently in use
- Substrate reduction therapy
- Molecular chaperone therapy - exogenous competitive inhibitor that binds mutant enzyme and acts as “folding template.” Tx under investigation for use in Gaucher disease
Tay-Sachs disease is most common form of GM2 gangliosidosis and results from mutations on what chromosome, leading to?
Prevalent in what population?
- α-subunit locus on chromosome 15 causing severe deficiency in lysosomal hexosaminidase A.
- Prevalent among Jews, paticularly Eastern European (Ashkenazic) origin
The enzyme deficiency in Tay-Sachs disease causes accumulation of GM2 gangliosides in many tissues, but which tissues dominate the clinical picture?
What is pictured with an electron microscope?
- Neurons in the central and autonomic nervous systems and retina
- Cytoplasmic inclusions, the most prominent being whorled configurations within lysosomes composed of onion-skin layers of membrane

A cherry-red spot appearing in the macula is characteristic of?
Tay-Sachs disease and other storage disorders affecting the neurons
Infants with Tay-Sachs disease begin to manifest signs and symptoms when?
What type of signs and symtoms?
- 6 months of age
- Relentless motor and mental deterioration, beginning with incoordination, obtundation, and muscular flaccidity
- Characteristic, but not pathognomonic cherry-red spot appears in macula of eye in almost all patients
- Complete vegetative state by 1-2 years, death by age 2-3
The gene mutation in Tay-Sachs disease leads to?
Misfolded protein –> unfolded protein response/ER stress response –> apoptosis
*Possibility of future “chaperone therapy” to treat this diseas
Niemann-Pick disease types A and B are characterized by lysosomal accumulation of?
Gene for defective enzyme map to which chromosome?
Preferentially expressed from maternal or paternal chromosome?
- Sphingomyelin due to inherited deficiency of sphingomyelinase
- 11p15.4
- Preferentially expressed from the maternal chromosome as result of epigenetic silencing of the paternal gene
Niemann-Pick disease types A and B are commonly seen in what population?
Ashkenazi Jews
Explain the pathogensis and clinical manifestations of Niemann-Pick disease type A?
Type of mutation?
What is seen in these patients and what is the prognosis?
- Severe infantile form with extensive neurological involvement, marked visceral accumulations of sphingomyelin, and progressive wasting
- Missense mutation causes almost complete deficiency of sphingomyelinase
- May be present at birth and almost invariably become evident by age 6 months, infants have protruberant abdomen because of the hepatosplenomegaly
- Death usually within the first or second year of life
What constitutes the dominant histological change seen in Niemann-Pick type A?
- Vacuolation and ballooing of neurons
- Concentric lamellated myelin figures called zebra bodies

Explain the pathogensis of Niemann-Pick disease type B?
Prognosis?
- Patients have organomegally but generally NO CNS involvement
- Usually survive into adulthood
What are the features of the brain like with someone who has Niemann-Pick type A?
Gyri are shrunken and sulci widened
How is the diagnosis of Niemann-Pick disease established?
Biochemical assays for sphingomyelinase activity in liver or bone marrow biopsy
Niemann-Pick disease type C is due to a primary defect in?
Mutations in what genes?
- Mutations in NPC1 (membrane bound) and/or NPC2 (soluble); both involved in transport of free cholesterol from the lysosomes to the cytoplasm
- NPC1 is responsible for 95% of cases
- Causes primary defect in NON-enzymatic lipid transport
How may Niemann-Pick type C present?
- Hydrops fetalis and stillbirth
- Neonatal hepatitis
- Most commonly as chronic form characterized by progressive neuro damage; presents in childhood and is marked by ataxia, vertical supranuclear gaze palsy, dystonia, dysarthria, and psychomotor regression
What is the most common lysosomal storage disoder and what is the affected gene?
- Gaucher disease
- Cluster of AR disorders from mutation in the gene encoding glucocerebrosidase (cleaves glucose from ceramide)
Glucocerebrosides are continually formed from the catabolism of?
Glycolipids derived mainly from the cell membranes of senescent leukocytes and red cells
Pathologic changes of Gaucher disease are caused not just by the burder of storage material but also by the activation and secretion of?
Activation of macrophages and secretion of cytokines such as IL-1, IL-6, and TNF
What is the most common form of Gaucher disease?
Affects primarily?
Found prinicipally in what population?
Affect on longevity?
- Type I, or the chronic nonneuronopathic form
- Storage of glucocerebrosides is limited to the mononuclear phagocytes throughout body WITHOUT involving the brain
- Splenic and skeletal involvements dominate this pattern
- Found principally in Jews of European stock
- Have reduced but detectable levels of glucocerebrosidase activity; longevity is shortened, but no markedly
Type II form of Gaucher disease has what type of affect and pattern?
Glucocerebrosidase activity?
Clinical picture dominated by?
Affect on longevity?
- Acute neuronopathic form, is the infantile acute cerebral pattern
- Virually no glucocerebrosidase activity in the tissues
- Hepatosplenomegaly seen, but clinical picture dominated by progressive CNS involvement, leading to death at an early age
What is the morphology of Gaucher disease?
Dominant cell type visualized?
What is visualized with electron microscope?
What type of stain is positive?
- Accumulation of phagocytotic cells, known as Gaucher cells, found in spleen, liver, bone marrow, LN’s, tonsils, thymus, and Peyers pathces
- Rarely appear vacuolated but instead have fibrillary type of cytoplasm likened to crumpled tissue paper
- Periodid acid-Schiff staining is intensely positive

Accumulation of Gaucher cells in the bone marrow in type I disease leads to?
- Areas of bone erosion, which can give rise to pathologic fractures
- Bone destruction occurs due to the secretion of cytokines (IL-1, IL-6, and TNF) by activated macrophages
The signs and symptoms of type I Gaucher disease first appear when and what’s involved?
Most commonly there is?
Longevity of these patients?
- In adult life and are related to splenomegaly or bone involvement
- Most commonly there is panocytopenia or thrombocytopenia secondary to hypersplenism
- Progressive in the adult, but IS compatible with long life
In types II and III Gaucher disease what are the most common dysfunctions and organs involved?
- CNS dysfunction, convulsions, and progressive mental deterioration dominate
- Liver, spleen and LN’s are also affected
The diagnosis of homozygotes with Gaucer disease can be made how?
Measuring glucocerebrosidase activity in peripheral blood leukocytes or in extracts of cultured skin fibroblasts
What is the mainstay treatment for Gaucher disease?
Effectivness and cost?
- Replacement therapy with recombinant enzymes
- Effective and those with type I can expect normal life expectancy
- Extremely expensive
- Allogenic hematopoietic stem cells transplantation can be curative
What are the cause of Mucopolysaccharidoses (MPSs)?
What are mucopolysaccharides and where are they most abundant?
- Deficiencies of enzymes that are involved in the degradation of mucopolysaccharides (glycosaminoglycans)
- Long-chain complex CHOs linked with proteins to form proteoglycans and are abundant in the ground substance of CT
What are the glycosaminoglycans that accumulate in MPSs?
Dermatan sulfate, heparan sulfate, keratan sulfate, and chondroitin sulfate
All the MPSs are classified numerically MPS I to MPS VII and are inherited in a _______ fashion
What is the exception?
- AR fashion
- Exception, Hunter syndrome, is X-linked recessive trait
In general, MPSs are progrssive disorders characterized by?
Coarse facial features, clouding of the cornea, joint stiffness, and menal retardation
Hepatosplenomegaly, skeletal deformities, valvular lesions, and subendothelial arterial deposits, particularly in the coronary arteries, and lesions in the brain are common threads that run through all forms of which disease?
MPSs
Hurler syndrome (MPS I-H) results from defiency of?
Appears when and what are its affects?
What is the cause of death?
- α-1-iduronidase deficiency
- One of the most severe forms and affected children develop hepatosplenomegaly by age 6 to 24 months
- Growth is retarded and develop coarse facial features and skeletal deformities
- Death occyrs by age 6-10 years, often due to cardiovascular complication
Hunter syndrome (MPS II) differs from Hurler syndrome how?
Mode of inheritance (X-linked), absence of corneal clouding and milder clinical course
MPSs have what feature that is different from all other storage disorders?
Metabolite present on urinalysis
What are the important causes of death seen in the prolonged forms of MPSs?
Coronary subendothelial lesions lead to myocardial ischemia, thus MI and cardiac decompensation contribute to death
Glucose molecules in glycogen are linked together via what kind of bonds?
α-1,4-glucoside bonds
Degradation by phosphorylases in liver and muscle split glucose-1-phosphate from glycogen until about 4 glucose residues remain on each branch, leaving a branch called?
How can this be further degraded?
- Limit dextrin
- Further degraded only by debranching enzyme
Von Gierke disease (type I glycogenosis) is deficiency in?
Characterized by?
Longevity?
- Glucose-6-phosphatase
- Hepatomegaly, renomegaly, impaired gluconeogenesis leading to hypoglycemia, hyperlipidemia, and hyperuricemia
- Most survive and develop late complications (i.e., hepatic adenomas)
What are the myopathic types of the glycogen storage diseases; enzymes that are deficient?
Clinical features of this form?
Onset when?
- McArdle disease (type V) = Muscle phosphorylase deficiency
- Type VII glycogen storage disease = defect in PFK
- Onset in adulthood (>20 year); muscle cramps after exercise and lactate levels in the blood fail to rise after exercise to due a block in glycolysis
- Serum creatine kinase always elevated
In Von Gierke disease where does the glycogen accumulate in the liver and kidney?
- Intracytoplasmic and intranuclear accumulations in liver
- Intracytoplasmic accumulation in cortical tubular epithelial cells of kidney
Pompe disease (type II glycogenosis) is a deficiency in?
Clinical features of this disease and what is the most prominent clinical feature?
- Lysosomal α-glucosidase (acid maltase)
- Lysosomal storage of glycogen in all organs (i.e., mild hepatomegaly and skeletal muscle) but massive cardiomegaly is most prominent feature
- Massive cardiomegaly, muscle hypotonia, and cardiorespiratory failure within 2 years of life
Describe the picture on the left vs. the one on the right

Left = normal myocardium with adundant eosinophilic cytoplasm
Right = patient with Pompe disease showing myocardial fibers full of glycogen seen as clear spaces

Which stain is most commonly used when identifying individual chromosomes?
Giemsa stain and is called G banding; usual procedure is to arrest in metaphase w/ mitotic spindle inhibitors
The notation Xp21.2 refers to?
Chromosomal segment located on short arm of X chromosome, in region 2, band 1, and sub-band 2
Euploid vs. aneuploidy?
Euploid = exact multiple of the haploid number of chromosomes (23)
Aneuploidy = error in mitosis or meiosis where cell acquires a chromosome complement that is not an exact multiple of 23
Usualy causes of aneuploidy are?
Nondisjunction and anaphase lag
Mosaicism is a result of and what occurs?
Most typically involves which chromosomes?
- Mitotic errors in early development give rise to 2 or more cell populations w/ different chromosomal complement, in the same individual
- Most commonly affects sex chromosomes; involvement of an autosome almost always leads to a nonviable mosaic
Which system allows for the detection of chromosomal changes as small as kilobases?
Fluoresence in situ hybridization (FISH)
46,XY,del(16)(p11.2p13.1) describes what kind of deletion?
Describes breakpoints in the short arm of chromosome 16 at 16p11.2 and 16p13.1 with loss of material between breaks
What is the most common example of isochromosome present in live births?
What is the Xq isochromosome for short arm and long arms genes?
- Involves long arm of the X and is designated i(X)(q10)
- Xq isochromosome is associated with monosomy for genes on short arm of X and with trisomy for genes on the long arm of X
What is a Robertsonian translocaton (or centric fusion)?
Where do the breaks most often occur and the end result?
Is it phenotypically compatible?
- Translocation between two acrocentric chromosomes
- Breaks occur close to the centromeres of each chromosome and transfer of segments lead to one very large and one extrememly small chromosome
- Small product usually lost; however carries highly redundant genes (i.e., ribosomal RNA genes), thus loss is compatible with normal phenotype
What is the most common manifestation of dyslipidemia?
Most commonly associated with what?
- Xanthomas
- High levels of LDL
Metabolic block and decreased amount of end product is associated with what?
Lesch-Nyhan
What is the most common cause of trisomy (Down syndrome)?
Age of which parent has strong influence on the incidence of Trisomy 21?
- Meiotic nondisjunction
- Maternal age; increase in age = increased incidence
What are the 3 genetic causes of Down’s syndrome?
- 95% have extra chromosome
- 4% Robertsonian translocation
- 1% are Mosaics
Congenital heart defects are common in patients with Down’s Syndrome, most commonly seen as defects of?
- Endocardial cushions, including:
- Ostium primum
- Atrial Septal defects
- AV valve malformation
- Ventricular septal defects
What is responsible for a majority of the deaths in infancy and early adulthood in those with Down’s Syndrome?
Cardiac problems
Patients with Down’s syndrome are at risk for what?
- Congenital heart disease
- Acute Leukemia (both acute lymphoblastic and acute myeloid) w/ acute megalaryoblastic leukima the most common
- Neuropathologic changes characteristic of Alzheimers by age 40
- Serious infections particularly of the lungs
What are the distinctive clinical features of Trisomy 18: Edwards syndrome?
- Prominent occiput
- Horseshoe kidney
- Low set ear
- Short neck
- Overlapping fingers
- Limited hip abduction

What is the basis for the emerging powerful noninvasive method of testing maternal blood used in the prenatal diagnosis of trisomy 21?
5-10% of total cell free DNA in maternal blood is derived from the fetus and can be identified by polymorphic genetic markers
What are the distinctive clinical features of trisomy 13: Patau syndrome?
- Micropthalmia (small or missing eyeballs)
- Polydactyly
- Microcephaly

What are the clinical features of DiGeorge’s syndrome?
- Thymic hypoplasia w/ resultant T-cell immunodeficiency
- Parathyroid hypoplasia giving rise to hypocalcemia
- Low-set ears, wide-set eyes, small jaw
What 2 genes are implicated in 22q11.2 deletion syndrome?
- TBX1 is most closely associated w/ the phenotypic features; also regulates PAX9
- PAX9 controls development of the palate, parathyroids, and thymus
What is the only consistent finding in Klinefelter syndrome (XXY)?
Hypogonadism
Most patients with Kleinfelter syndrome have a distinctive body habitus characterized by?
- Increase in length between the soles and the pubic bone = elongated appearance
- Abnormally long legs
- Small testes and penis
- Lack of secondary sexual characteristics: deep voice, beard
Patients with Klinefelter syndrome are at higher risk for what?
- Breast cancer
- Type 2 diabetes (insulin resistance)
- Mitral valve prolapse
Where does the androgen receptor gene map to and what does it contain?
How does this contribute to hypogonadism observed in patients with Kleinfelter Syndrome?
- Mapped to the X chromosome and contains highly polymorphic CAG (trinucleotide) repeats
- Receptors with shorter CAG repeats are more sensitive to androgens
- In Klinefelters, the X chromosome bearing the androgen receptor allele w/ the shortest CAG repeat is preferentially silenced, thus the remaining X chromosome w/ the androgen receptor allele containing more CAG repeats is expressed
Severely affected patients with Turner Syndrome are born with?
- Edema of the dorsum of hand and foot (lymph stasis)
- Swelling of the nape of neck (cystic hygroma)
- Swelling subside and leave bilateral neck webbing and persisten looseness of skin on back of the neck
Which heart abnormalities are seen most frequently in patients with Turner’s Syndrome?
- Left-sided cardiovascular abnormalities
- Pre-ductal coarctation of the aorta and bicuspid aortic valve
* Most important causes of increased mortality in children w/ Turner’s
What are the distinguishing clinical features of Turner’s Syndrome?
- Short stature
- Amenorrhea (single most important cause of primary amenorrhea)
- Streak ovaries
- Cystic hygroma of the neck and hydrops fetalis
- Cubitus Valgus

Single most important cause of primary amenorrhea?
Turner syndrome
What leads to the developement of streak ovaries in patients with Turner’s Syndrome?
- Fetal ovaries develop normally in embryogenesis, but the absence of the second X chromosome leads to an accelerated loss of oocytes, which is complete by age 2
- “Menopause occurs before menarch.”
- Ovaries are reduced to atrophic fibrous stands, devoid of ova and follicle
Which gene is associated with the short-stature seen in patients with Turner’s syndrome?
Haploinsufficiency of SHOX gene at Xp22.33
Fragile-X syndrome is a result of what?
What type of mutation?
- CGG tricnucleotide repeats (non-coding part of gene)
- Loss of function
What is the morphologic hallmark of polyglutamine expansions in the coding regions of genes that lead to toxic gain of function diseases?
Accumulation of aggregated mutant proteins in large intranuclear inclusions
Toxic gain of function mediated by mRNA where there are expansions in noncoding parts of the gene lead to what disease?
Fragile X Tremor-Ataxia Syndrome = toxic gain of function of FMR1 gene
Fragile X syndrome is caused by mutation in what gene?
2nd most common genetic cause of?
- FMR1 gene
- Mental retardation
What is the characteristic phenotype of someone with Fragile X syndrome?
- Long face w/ large manible
- Large everted ears
- Large testicles (macro-orchidism) = most distinctive feature
When the trinucleotide repeats in the FMR1 gene exceed 230, what occurs to the DNA of the entire 5’ region of the gene?
- Becomes abnormally methylated
- Methylation also extends upstream into the promoter region of the gene, resulting in transcriptional suppression of FMR1
Where is the FMRP protein found most abundantly?
What are its functions?
- Brain and Testis
- Selectively binds and regulates the transport of mRNA into dendrites
- At synaptic junctions FMRP suppresses protein synthesis of the bound mRNAs in response to signaling through group I metabotropic glutamate receptors (mGlu-R) = translation regulator
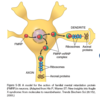
What is the neurodegenerative disease that manifests as progressive bilateral loss of central vision, first notes between ages 15-13 and eventually causes blindness?
Type of inheritance?
- Leber hereditary optic neuropathy
- Mitochondrial inheritance
What is genomic imprinting?
Paternal vs. maternal imprinting?
- We inherit 2 copies of each autosomal gene, carried on homologous maternal and paternal chromosomes
- Imprinting is an epigenetic process, in which either the maternal or paternal allele is selectively inactivated
- Maternal imprinting is transcriptional silencing of the maternal allele, while paternal imprinting is silencing of the paternal allele
What syndrome is characterized by mental retardation, short stature, hypotonia, profound, hyperphagia, obesity, small hands and feet, hypogonadism?
What is the underlying cause of this disease?
- Prader-Willi syndrome
- Deletion affecting the paternally derived chromosome 15 (specifically deletion of band q12 in long arm of chromosome 15)
- Only the functional allele is provided by the paternal chromosome

Mental retardation, ataxic gait, seizures and innapropriate laughter (“happy puppets”) describes what disorder?
Caused by what gene and process?
- Angelman syndrome
- UBE3A gene mapped to maternal chromosome 15 is deleted. This gene is imprinted on the paternal allele, but only the maternal allel is normally active

Which family of genes located in 15q11.2-q13 are believed to be involved in Prader-Willi syndrome?
Loss of the SNORP family of genes that encode small nucleolar RNAs involved in the modifications of ribosomal RNAs
Cystic fibrosis is due to what genetic mutation?
Which type of transmission pattern?
- 3 base deletion in CF allele, resulting in lack of AA508 (Phe) on chromosome 7
- Autosomal recessive
How is an Autosomal Dominant disorder, such as Osteogenesis Imperfecta, arising from phenotypically normal parents able to affect more than one child?
What is the mechanism?
- Gonadal Mosaicism
- Mutations that occur postzygotically during early (embryonic) development that affects only cells to be the gonads.
- Gametes carry the mutation, but the somatic cells of the individual are completely normal; so the disease-causing mutation is transmitted to the offspring through the mutated gametes
What are the 5 mutation classes for familial hypercholesterolemia? (mnemonic)
Some Times Bitches Come Real
1 - Synthesis
2 - Transport
3 - Binding
4 - Clustering
5 - Recycling
What disease microscopically has the appearance of balloon cells in the cytoplasm?
MPS
Upon electron microscopy there is presence of vacuoles engorged secondary lysosomes containing membranous cytoplasmic bodies, resembling concentric lamellated myelin figures, called zebra bodies, what does this indicate?
Niemann-Pick Type A
What is the trinucleotide repeat for Huntington disease?
CAG
Upon exam with electron microscope, the presence of whorled configurations within lysosomes composed of onion-skin layers of membranes indicates what?
Tay-Sachs
What disease is charcterized by a fibrillary type of cytoplasm likened to crumpled tissue paper?
Gaucher
What is the trinucleotide repeat for Friedreich ataxia?
GAA
Enzymes deficient in Von Gierke disease (type I)?
McArdle disease (type V)?
Pompe disease (type II)?
- Von Gierke = glucose-6-phosphatase
- McArdle = muscle phosphorylase
- Pompe = α-glucosidase (acid maltase)
What is the trinucleotide repeat for Myotonic dystrophy?
CTG
Mode of inheritance for Hunter Syndrome vs. Hurler Syndrome?
Hunter = X-linked recessive
Hurler = autosomal recessive
Mitral valve prolapse (AKA floppy valve) and dilation of the ascending aorta are associated with what condition?
Marfan Syndrome
