Chapter 23. Protein Turnover and Amino Acid Catabolism Flashcards
Question 23.1
Getting exposure. Proteins are denatured by acid in the stomach. This denaturation makes them better substrates for proteolysis. Explain why this is the case.
- When the proteins are denatured, all of the peptide bonds are accessible to proteolytic enzymes. If the three-dimensional structure of a protein is maintained, access to many peptide bonds is denied to the proteolytic enzymes.
Question 23.2
Targeting for destruction. What are the steps required to attach ubiquitin to a target protein?
- First, the ubiquitin-activating enzyme (E1) links ubiquitin to a sulfhydryl group on E1 itself. Next, the ubiquitin is transferred to a cysteine residue on the ubiquitin-conjugating enzyme (E2) by E2. The ubiquitin–protein ligase (E3), using the ubiquitinated E2 as a substrate, transfers the ubiquitin to the target protein.
Question 23.4
Wasted energy? Protein hydrolysis is an exergonic process, yet the 26S proteasome is dependent on ATP hydrolysis for activity.
Explain why ATP hydrolysis is required by the 26S proteasome.
Small peptides can be hydrolyzed without the expenditure of ATP. How does this information concur with your answer to part a?
- (a) The ATPase activity of the 26S proteasome resides in the 19S subunit. The energy of ATP hydrolysis is used to unfold the substrate, which is too large to enter the catalytic barrel. ATP may also be required for translocation of the substrate into the barrel.
(b) Substantiates the answer in part a. Because they are small, the peptides do not need to be unfolded. Moreover, small peptides could probably enter all at once and not require translocation.
Question 23.5
Keto counterparts. Name the α-ketoacid that is formed by the transamination of each of the following amino acids:
- Alanine
- Aspartate
- Glutamate
- Leucine
- Phenylalanine
- Tyrosine
- (a) Pyruvate; (b) oxaloacetate; (c) α-ketoglutarate; (d) α-ketoisocaproate; (e) phenylpyruvate; (f ) hydroxyphenylpyruvate.
Question 23.6
A versatile building block.
Write a balanced equation for the conversion of aspartate into glucose through the intermediate oxaloacetate. Which coenzymes participate in this transformation?
Write a balanced equation for the conversion of aspartate into oxaloacetate through the intermediate fumarate.
- (a) Aspartate + α-ketoglutarate + GTP + ATP + 2 H2O + NADH + H+ → ½ glucose + glutamate + CO2 + ADP + GDP + NAD+ + 2 Pi.
The required coenzymes are pyridoxal phosphate in the transamination reaction and NAD+/NADH in the redox reactions.
(b) Aspartate + CO2 + NH4+ + 3 ATP + NAD+ + 4 H2O → oxaloacetate + urea + 2 ADP + 4 Pi + AMP + NADH + H+.
Question 23.7
The benefits of specialization. The archaeal proteasome contains 14 identical active β subunits, whereas the eukaryotic proteasome has 7 distinct β subunits. What are the potential benefits of having several distinct active subunits?
- In the eukaryotic proteasome, the distinct β subunits have different substrate specificities, allowing proteins to be more thoroughly degraded.
Question 23.10
Cooperation. How do aminotransferases and glutamate dehydrogenase cooperate in the metabolism of the amino group of amino acids?
- Aminotransferases transfer the α-amino group to α-ketoglutarate to form glutamate. Glutamate is oxidatively deaminated to form an ammonium ion.
Question 23.11
Taking away the nitrogen. What amino acids yield citric acid cycle components and glycolysis intermediates when deaminated?
- Aspartate (oxaloacetate), glutamate (α-ketoglutarate), alanine (pyruvate).
Question 23.12
One reaction only. What amino acids can be deaminated directly?
- Serine and threonine.
Question 23.13
Useful products. What are the common features of the breakdown products of the carbon skeletons of amino acids?
- They are either fuels for the citric acid cycle, components of the citric acid cycle, or molecules that can be converted into a fuel for the citric acid cycle in one step.
Question 23.15
Nitrogen sources. What are the immediate biochemical sources for the two nitrogen atoms in urea?
- Carbamoyl phosphate and aspartate.
Question 23.18
Completing the cycle. Four high-transfer-potential phosphoryl groups are consumed in the synthesis of urea according to the stoichiometry given in Section 23.4. In this reaction, aspartate is converted into fumarate. Suppose that fumarate is converted into oxaloacetate. What is the resulting stoichiometry of urea synthesis? How many high-transfer-potential phosphoryl groups are spent?
18.
Four high-transfer-potential phosphoryl groups are spent. Note, however, that an NADH is generated if fumarate is converted into oxaloacetate. NADH can generate 2.5 ATP in the electron-transport chain. Taking these ATP into account, only 1.5 high-transfer-potential phosphoryl groups are spent.
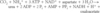
Question 23.19
A good bet. A friend bets you a bazillion dollars that you can’t prove that the urea cycle is linked to the citric acid cycle and other metabolic pathways. Can you collect?
- The synthesis of fumarate by the urea cycle is important because it links the urea cycle and the citric acid cycle. Fumarate is hydrated to malate, which, in turn, is oxidized to oxaloacetate. Oxaloacetate has several possible fates: (1) transamination to aspartate, (2) conversion into glucose by the gluconeogenic pathway, (3) condensation with acetyl CoA to form citrate, or (4) conversion into pyruvate. You can collect.
Question 23.21
Ammonia toxicity. Glutamate is an important neurotransmitter whose levels must be carefully regulated in the brain. Explain how a high concentration of ammonia might disrupt this regulation. How might a high concentration of ammonia alter the citric acid cycle?
- Ammonia could lead to the amination of α-ketoglutarate, producing a high concentration of glutamate in an unregulated fashion. α-Ketoglutarate for glutamate synthesis could be removed from the citric acid cycle, thereby diminishing the cell’s respiration capacity.
Question 23.22
A precise diagnosis. The urine of an infant gives a positive reaction with 2,4-dinitrophenylhydrazine. Mass spectrometry shows abnormally high blood levels of pyruvate, α-ketoglutarate, and the α-ketoacids of valine, isoleucine, and leucine. Identify a likely molecular defect and propose a definitive test of your diagnosis.
- The mass spectrometric analysis strongly suggests that three enzymes—pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, and the branched-chain α-ketoacid dehydrogenase—are deficient. Most likely, the common E3 component of these enzymes is missing or defective. This proposal could be tested by purifying these three enzymes and assaying their ability to catalyze the regeneration of lipoamide.
Question 23.23
Therapeutic design. How would you treat an infant who is deficient in argininosuccinate synthetase? Which molecules would carry nitrogen out of the body?
- Benzoate, phenylacetate, and arginine would be given to supplement a protein-restricted diet. Nitrogen would emerge in hippurate, phenylacetylglutamine, and citrulline.
Question 23.24
Damaged liver. As we will see later (Chapter 27), liver damage (cirrhosis) often results in ammonia poisoning. Explain why this is the case.
- The liver is the primary tissue for capturing nitrogen as urea. If the liver is damaged (for instance, by hepatitis or the excessive consumption of alcohol), free ammonia is released into the blood.
Question 23.25
Argininosuccinic aciduria. Argininosuccinic aciduria is a condition that results when the urea-cycle enzyme argininosuccinase is deficient. Argininosuccinate is present in the blood and urine. Suggest how this condition might be treated while still removing nitrogen from the body.
- This defect can be partly bypassed by providing a surplus of arginine in the diet and restricting the total protein intake. In the liver, arginine is split into urea and ornithine, which then reacts with carbamoyl phosphate to form citrulline. This urea-cycle intermediate condenses with aspartate to yield argininosuccinate, which is then excreted. Note that two nitrogen atoms—one from carbamoyl phosphate and the other from aspartate—are eliminated from the body per molecule of arginine provided in the diet. In essence, argininosuccinate substitutes for urea in carrying nitrogen out of the body. The formation of argininosuccinate removes the nitrogen, and the restriction on protein intake relieves the aciduria.
Question 23.26
Sweet hazard. Why should phenylketonurics avoid using aspartame, an artificial sweetener? (Hint: Aspartame is l-aspartyl-l-phenylalanine methyl ester.)
- Aspartame, a dipeptide ester (l-aspartyl-l-phenylalanine methyl ester), is hydrolyzed to l-aspartate and l-phenylalanine. High levels of phenylalanine are harmful in phenylketonurics.
Question 23.28
Negative nitrogen balance. A deficiency of even one amino acid results in a negative nitrogen balance. In this state, more protein is degraded than is synthesized, and so more nitrogen is excreted than is ingested. Why would protein be degraded if one amino acid were missing?
- Not all proteins are created equal: some are more important than others. Some proteins would be degraded to provide the missing amino acid. The nitrogen from the other amino acids would be excreted as urea. Consequently, more nitrogen would be excreted than ingested.
Question 23.29
Precursors. Differentiate between ketogenic amino acids and glucogenic amino acids.
- The carbon skeletons of ketogenic amino acids can be converted into ketone bodies or fatty acids. Only leucine and lysine are purely ketogenic. Glucogenic amino acids are those whose carbon skeletons can be converted into glucose.
Question 23.30
A slight of hand. The end products of tryptophan degradation are acetyl CoA and acetoacetyl CoA, yet tryptophan is a gluconeogenic amino acid in animals. Explain.
- As shown in Figure 23.28, alanine, a gluconeogenic amino acid, is released during the metabolism of tryptophan to acetyl CoA and acetoacetyl CoA.
Question 23.31
Closely related. Pyruvate dehydrogenase complex and α-ketoglutarate dehydrogenase complex are huge enzymes consisting of three discrete enzymatic activities. Which amino acids require a related enzyme complex, and what is the name of the enzyme?
- The branched-chain amino acids leucine, isoleucine, and valine. The required enzyme is the branched-chain α-ketoacid dehydrogenase complex.
Question 23.32
Supply lines. The carbon skeletons of the 20 common amino acids can be degraded into a limited number of end products. What are the end products and in what metabolic pathway are they commonly found?
- Pyruvate (glycolysis and gluconeogenesis), acetyl CoA (citric acid cycle and fatty acid synthesis), acetoacetyl CoA (ketone-body formation), α-ketoglutarate (citric acid cycle), succinyl CoA (citric acid cycle), fumarate (citric acid cycle), and oxaloacetate (citric acid cycle and gluconeogenesis).
Question 23.35
Multiple substrates. In Chapter 8, we learned that there are two types of bisubstrate reactions, sequential and double-displacement. Which type characterizes the action of ami-notransferases? Explain your answer.
- Double-displacement. A substituted enzyme intermediate is formed.
Question 23.36
Double duty. Degradation signals are commonly located in protein regions that also facilitate protein–protein interactions. Explain why this coexistence of two functions in the same domain might be useful.
- Exposure of such a domain suggests that a component of a multiprotein complex has failed to form properly or that one component has been synthesized in excess. This exposure leads to rapid degradation and the restoration of appropriate stoichiometries.
Question 23.37
Fuel choice. Within a few days after a fast begins, nitrogen excretion accelerates to a higher-than-normal level. After a few weeks, the rate of nitrogen excretion falls to a lower level and continues at this low rate. However, after the fat stores have been depleted, nitrogen excretion rises to a high level.
What events trigger the initial surge of nitrogen excretion?
Why does nitrogen excretion fall after several weeks of fasting?
Explain the increase in nitrogen excretion when the lipid stores have been depleted.
- (a) Depletion of glycogen stores. When they are gone, proteins must be degraded to meet the glucose needs of the brain. The resulting amino acids are deaminated, and the nitrogen atoms are excreted as urea.
(b) The brain has adapted to the use of ketone bodies, which are derived from fatty acid catabolism. In other words, the brain is being powered by fatty acid breakdown.
(c) When the glycogen and lipid stores are gone, the only available energy source is protein.
Question 23.38
A serious situation. Pyruvate carboxylase deficiency is a fatal disorder. Patients with pyruvate carboxylase deficiency sometimes display some or all of the following symptoms: lactic acidosis, hyperammonemia (excess NH4+ in the blood), hypoglycemia, and demyelination of the regions of the brain due to insufficient lipid synthesis. Provide a possible biochemical rationale for each of these observations.
- The precise cause of all of the symptoms is not firmly established, but a likely explanation depends on the centrality of oxaloacetate to metabolism. A lack of pyruvate carboxylase would reduce the amount of oxaloacetate. The lack of oxaloacetate would reduce the activity of the citric acid cycle and so ATP would be generated by lactic acid formation. If the concentration of oxaloacetate is low, aspartate cannot be formed and the urea cycle would be compromised. Oxaloacetate is also required to form citrate, which transports acetyl CoA to the cytoplasm for fatty acid synthesis. Finally, oxaloacetate is required for gluconeogenesis.
