Chapter 18 –Metabolic Liver Disease - α1-ANTITRYPSIN DEFICIENCY Flashcards
What is α1-Antitrypsin deficiency?
- autosomal recessive disorder
- marked by very low levels of α1- antitrypsin.
What is the major function of α1-Antitrypsin?
The major function of this protein is the inhibition of proteases, particularly neutrophil elastase, cathepsin G, and proteinase 3, which are normally released from neutrophils at sites of inflammation.
What happens when there is α1-Antitrypsin deficiency?
leads to the development of pulmonary emphysema, because the activity of destructive proteases is not inhibited (discussed in Chapter 15 ).
It also causes liver disease, as a consequence of the accumulation of this protein in hepatocytes. [52]
In addition, cutaneous panniculitis, arterial aneurysm,
bronchiectasis, and Wegener’s granulomatosis can occur in α1-antitrypsin deficiency.
Describe α1-Antitrypsin.
- small 394–amino acid plasma glycoprotein
- synthesized predominantly by hepatocytes.
- It is a member of the serine protease inhibitor (serpin) family.
- located on chromosome 14, is very polymorphic, and at least 75 α1-antitrypsin forms have been identified, denoted alphabetically by their relative migration on an isoelectric gel.
The general notation is “Pi” for “protease inhibitor” and an alphabetic letter for the position on the gel;
two letters denote the genotype of the two alleles.
What is the wild-type genotype of α1-
antitrypsin?
The most common genotype is PiMM, occurring
in 90% of individuals (in the traditional sense, this would be the wild-type genotype).
Most allelic
variants show substitutions in the polypeptide chain but produce normal levels of functional α1-
antitrypsin.
Some deficiency variants, including the PiS variant, result in a moderate reduction in
serum concentrations of α1-antitrypsin without clinical manifestations.
Rare variants termed Pinull
have no detectable serum α1-antitrypsin.
What is the most common clinically significant mutation in the α1-antitrypsin deficiency?
PiZ;
homozygotes for the PiZZ protein have _circulating α1-antitrypsin levels that are only 10% of
normal._
These individuals are at high risk for developing clinical disease.
Expression of alleles is autosomal codominant, and consequently, PiMZ heterozygotes have intermediate plasma levels of α1-antitrypsin.
Among people of northern European descent the PiS frequency is 6% and the PiZ frequency is 4%; the PiZZ state affects 1 in 1800 live births.
Why is α1-antitrypsin deficiency is the most commonly
diagnosed genetic hepatic disorder in infants and children?
Because of its
occasionally early presentation for liver disease.
What is the pathogenesis of α1-antitrypsin deficiency?
With most allelic variants, the mRNA is transcribed, and the protein is synthesized and secreted
normally.
Deficiency variants show a selective defect in migration of this secretory protein from the endoplasmic reticulum to Golgi apparatus; this is most marked for the PiZ polypeptide, attributable to a single amino acid substitution of Glu342 to Lys342.
The mutant polypeptide
( α1AT-Z) is abnormally folded and polymerizes , creating endoplasmic reticulum stress and
leading to apoptosis ( Chapter 1 ; see Fig. 1-27 ).
The precise mechanisms of liver disease with
α1AT-Z are not well defined.
The accumulated α1AT-Z in the endoplasmic reticulum triggers a series of events, including an autophagocytic response, mitochondrial dysfunction, and possible activation of pro-inflammatory NF-κB, causing hepatocyte damage. [53]
All individuals with the PiZZ genotype accumulate α1AT-Z in the endoplasmic reticulum of hepatocytes, but only 10% to 15% of PiZZ individuals develop overt clinical liver disease.
Other genetic factors or
environmental factors are thus posited to play a role in the development of liver disease.
What is the characteristic of α1-Antitrypsin deficiency?
is characterized by the presence of round-tooval
cytoplasmic globular inclusions in hepatocytes,*_which in routine _*H&E stains are
acidophilic and indistinctly demarcated from the surrounding cytoplasm.
They are
strongly periodic acid–Schiff (PAS)-positive and diastase-resistant ( Fig. 18-27 ).
The globules are also present but in diminished size and number in the PiMZ and PiSZ genotypes.
For unknown reasons most of the globules are in hepatocytes surrounding the portal tracts.
Moreover, the number of globule-containing hepatocytes in a patient’s liver is not correlated
with the severity of pathologic findings.
The hepatic pathology associated with PiZZ homozygosity is extremely varied, ranging from neonatal hepatitis ( Fig. 18-28 ) without or with cholestasis and fibrosis (discussed below), to childhood cirrhosis, to a smoldering chronic inflammatory hepatitis or cirrhosis that becomes apparent only late in life.
For the most part
the only distinctive feature of the hepatic disease is the PAS-positive globules; infrequently,
fatty change and Mallory bodies are present.
The diagnostic α1-antitrypsin globules may be
absent in the young infant; steatosis may be present as a tip-off to the possibility of α1- antitrypsin deficiency.

FIGURE 18-27 α1-Antitrypsin deficiency
. A, Periodic acid–Schiff (PAS) stain of the liver,
highlighting the characteristic red cytoplasmic granules.
B, Electron micrograph showing
the dilatation of the endoplasmic reticulum.
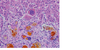
FIGURE 18-28 Neonatal hepatitis caused by α1-antitrypsin deficiency. Note the severe
cholestasis.
What are the clinical features of α1-ANTITRYPSIN DEFICIENCY?
Neonatal hepatitis with cholestatic jaundice appears in 10% to 20% of newborns with the deficiency.
In adolescence, presenting symptoms may be related to hepatitis or cirrhosis.
Attacks of hepatitis may subside with apparent complete recovery, or they may become chronic
and lead progressively to cirrhosis
. Finally, the disease may remain silent until cirrhosis appears in middle to later life.
HCC develops in 2% to 3% of PiZZ adults, usually but not always in the setting of cirrhosis.
Whais the treatment for α1-ANTITRYPSIN DEFICIENCY?
The treatment, and the cure, for severe hepatic disease is orthotopic liver transplantation.
In patients with pulmonary disease the single most important treatment is avoidance of cigarette smoking, because smoking markedly accelerates emphysema and the destructive lung disease associated with α1-antitrypsin deficiency
-END-
Chapter 18 –Metabolic Liver Disease - α1-ANTITRYPSIN DEFICIENCY
