Chapter 14 – Red Blood Cell: HEMOLYTIC ANEMIAS Flashcards
Hemolytic anemias share the following features
- • Premature destruction of red cells and a shortened red cell life span below the normal 120 days
- • Elevated erythropoietin levels and a compensatory increase in erythropoiesis
- • Accumulation of hemoglobin degradation products released by red cell breakdown derived from hemoglobin
Where does the physiologic destruction of senescent red cells takes place?
within mononuclear phagocytes,
which are abundant in the spleen, liver, and bone marrow.
What triggers the physiologic destruction of senescent red cells takes place?
age-dependent changes in red cell surface proteins, which lead to their recognition and
phagocytosis.
What is extravascular hemolysis?
[1] In the great majority of hemolytic anemias the premature destruction of red
cells also occurs within phagocytes, an event that is referred to as extravascular hemolysis.
What happens when there is a persistent extravascular hemolysis?
- *hyperplasia of phagocytes** manifested by varying
- *degrees of splenomegaly.**
What is the general caused of extravascular hemolysis?
Extravascular hemolysis is generally caused by alterations that render the red cell less
deformable.
What is required for RC to navigate the splenic sinusoids successfully?
Extreme changes in shape are required for red cells to navigate the splenic sinusoids successfully.
Reduced deformability makes this passage difficult, leading to red cell sequestration and phagocytosis within the cords
What makes the RC sequestration and phagocytosis within the cords?
Reduced deformability
makes this passage difficult, leading to red cell sequestration and phagocytosis within the cords
Regardless of the cause, the principal clinical
features of extravascular hemolysis are
- (1) anemia,
- (2) splenomegaly,
- and (3) jaundice
What is the reason for decrease in haptoglobin?
Some hemoglobin inevitably escapes from phagocytes, which leads to variable decreases in plasma
haptoglobin, an α2-globulin that binds free hemoglobin and prevents its excretion in the urine.
Why do individuals with extravascular hemolysis benefit form splenectomy?
Because much of the pathologic destruction of red cells occurs in the spleen, individuals with
extravascular hemolysis often benefit from splenectomy.
What is the cause of intravascular hemolysis?
Less commonly, intravascular hemolysis predominates.
Intravascular hemolysis of red cells may
be caused by mechanical injury, complement fixation, intracellular parasites (e.g., falciparum
malaria, Chapter 8 ), or exogenous toxic factors.
Causes of mechanical injury include trauma
caused by cardiac valves, thrombotic narrowing of the microcirculation, or repetitive physical
trauma (e.g., marathon running and bongo drum beating).
Complement fixation occurs in a
variety of situations in which antibodies recognize and bind red cell antigens.
Toxic injury is
exemplified by clostridial sepsis, which results in the release of enzymes that digest the red cell
membrane.
Whatever the mechanism, intravascular hemolysis is manifested by :
- (1) anemia,
- (2) hemoglobinemia,
- (3) hemoglobinuria,
- (4) hemosiderinuria, and
- (5) jaundice.
What is the reason for the red-brown color of urine in intravascular hemolysis?
The large amounts
of free hemoglobin released from lysed red cellsare promptlybound by haptoglobin, producing
a complex that is rapidly cleared by mononuclear phagocytes.
As serum haptoglobin is
depleted, free hemoglobinoxidizes to methemoglobin, which is brown in color.
The renal
proximal tubular cells reabsorb and catabolize much of the filtered hemoglobin and
methemoglobin,butsome passes out in the urine, imparting a red-brown color.
Iron released
from hemoglobin can accumulate within tubular cells, giving rise to renal hemosiderosis.
What is the reason for jaundice in intravascular hemolysis?
Concomitantly, heme groups derived from hemoglobinhaptoglobin complexes are catabolized to
bilirubin within mononuclear phagocytes, leading to jaundice.
Unlike in extravascular hemolysis,
________ is not seen intravascular hemolysis?
splenomegaly
In all types of uncomplicated hemolytic anemias, the excess serum bilirubin is ____________
unconjugated.
The level of hyperbilirubinemia depends on the functional capacity of the liver and the rate of
hemolysis. When the liver is normal, jaundice is rarely severe.
Excessive bilirubin excreted by
the liver into the gastrointestinal tract leads to increased formation and fecal excretion of
urobilin ( Chapter 18 ), and often leads to the formation of gallstones derived from heme pigments.
Certain changes are seen in hemolytic anemias regardless of cause or type.
Anemia and lowered tissue oxygen tension trigger the production of __________, which
stimulates erythroid differentiation and leads to the appearance of increased numbers of
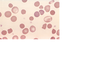
erythroid precursors (normoblasts) in the marrow ( Fig. 14-1 ).
erythropoietin
What results in the Compensatory
increases in erythropoiesis?
prominent reticulocytosis in the peripheral blood
What is hemosiderosis?
The phagocytosis of red cells leads to hemosiderosis, which is most pronounced in the
spleen, liver, and bone marrow.
Why does in chronic hemolysis formation of pigment gallstones can occur?
With chronic hemolysis, elevated biliary
excretion of bilirubin promotes the formation of pigment gallstones (cholelithiasis).

FIGURE 14-1 Marrow smear from a patient with hemolytic anemia. The marrow reveals
greatly increased numbers of maturing erythroid progenitors (normoblasts).
What is hereditary spherocytosis?
This inherited disorder is caused by intrinsic defects in the red cell membrane skeleton that
render red cells spheroid, less deformable, and vulnerable to splenic sequestration and destruction. [2]
The prevalence of HS is highest in northern Europe, where rates of 1 in 5000 are reported.
An autosomal dominant inheritance pattern is seen in about 75% of cases.
The
remaining patients have a more severe form of the disease that is usually caused by the
inheritance of two different defects (a state known as compound heterozygosity).
The remarkable elasticity and durability of the normal red cell are attributable to what?
The remarkable elasticity and durability of the normal red cell are attributable to the physicochemical properties of its specialized membrane skeleton ( Fig. 14-2 ), which lies closely apposed to the internal surface of the plasma membrane