Tubulointerstitial Disease: Nichols + Wall Flashcards
What are the lab indications for acute kidney injury? Categories?
- Serum creatinine: rise of at least 0.3 mg/dL over a 48-hr period and/or a rise ≥ 1.5 times the baseline value within the 7 previous days
- Urine volume: ≤0.5 mL/kg per hour for 6 hours

What are the causes of AKI? Long list.

What kinds of things can cause AKI via hemodynamic injury?
- ACEI, ARB (bilateral renal artery stenosis)
- Amphotericin B
- Calcineurin inhibitors
- Diuretics
- NSAIDs (bilateral renal artery stenosis)
- Radiocontrast
What kinds of things can cause AKI via thrombotic microangiopathy?
- Calcineurin inhibitors
- Clopidogrel
- Cocaine
- Mitomycin
- Quinine
What kinds of things can cause AKI via proximal tubule injury?
- Aminoglycosides
- Heavy metals: admium, lead, mercuric chloride
- Antiretrovirals: Acyclovir, Cidofovir, Tenofovir, Didanosine
- Cisplatin
- Foscarnet (antiviral for CMV)
What kinds of things can cause AKI via distal tubule injury?
- Amphotericin B
- Calcineurin inhibitors
- Lithium
What kinds of things can cause AKI via tubular obstruction?
- Acyclovir, Indinavir
- Methotrexate
- Sulfonamides
- CD: concentrating defects, obstruction, hyperkalemia
What kinds of things can cause AKI via interstitial nephritis?
- Allopurinol
- Aristolochic acid
- AB’s: Cephalosporins, Macrolides, Penicillins
- Ciprofloxacin
- Diuretics
- NSAID’s
- Phenytoin
- PPI’s
- NOTE: drug complications -> interstitial or tubular injury most common
What labs help you differentiate between pre-renal and intra-renal ATN?
- NOTE: hyaline casts are the only normal casts

What are the principle sites and causes of tubular injury in TIN?
- Different places of damage are going to cause different consequences
- Structure-function relationship is important

Acute Interstitial Nephritis

- Immune mediated hypersensitivity reaction to an antigen (e.g. drug or infection)
- Makes about 10-15% of all acute renal failure and about 1% of all renal biopsies done for hematuria and low grade proteinuria proteinuria
- NOT dose-dependent (idiosyncratic)
- Extra-renal manifestations of hypersensitivity: fever, skin rashes, arthralgias
- Recurrence with re-exposure
What do you see here?

Acute interstitial nephritis
Why is GFR impaired if the tubules are the ones that are damaged in AIN?
- INC pressure in the tubule, removing pressure gradient between capillary and tubule
- To have GFR, you have to have intact blood vessels, glomerulus, and tubules
- Tubular injury much more apt to screw up your homeostatic ability since they do all the work
What is the antigenic stimulus for AIN?
-
Antigens:
1. Tubular basement membrane
2. Secreted tubular proteins
3. Non-renal proteins (immune complexes) -
Immune activation via drugs or infectious agents:
1. As planted antigens
2. Acting as haptens to modify immunogenicity of native renal proteins
3. Molecular mimickery
4. Circulating immune complexes precipitation - NOTE: a hapten is a small molecule that can elicit an immune response only when attached to a large carrier such as a protein; the carrier may be one that also does not elicit an immune response by itself
What type of immune reaction is elicited in AIN?
- Cell-mediated immunity plays major role with activated T-cell infiltration and sometimes formation of granulomas
- Antibody mediated immunity may also play a role, especially in methicillin induced AIN
- Proliferation of interstitial fibroblast and matrix
- TGF-b plays critical role
What is the clinical presentation of AIN?
- Usually w/in 3 weeks of starting the new drug (or temporarily related to an infection)
- Sudden onset of renal insufficiency
- Fever, Rash, flank pain, hematuria, sterile pyuria (urine w/WBCs, pus), eosinophiluria
1. Bulk of WBC’s will be neutrophils - Minimal proteinuria -> glomerulus is fine
- Hemolysis, hepatitis (may also be present)
What are the common drugs associated with acute drug-induced interstitial nephritis?
- First reported after use of sulfonamides
-
Common drugs:
1. Synthetic penicillins (methicillin, ampicillin)
2. Rifampin, ciprofloxacin
3. Diuretics (thiazides)
4. NSAIDs
5. Allopurinol, Cimetidine, proton pump inhibitors - Disease begins about 15 days (range: 2–40) after exposure to the drug
- Triad of: fever, eosinophilia (which may be transient), a rash in 25%
What renal abnormalities are associated with acute drug-induced interstitial nephritis?
- Hematuria, typically microscopic
- Minimal proteinuria
- Leukocyturia (often including eosinophiluria)
- A rising serum creatinine level (because GFR going down) or acute renal failure with oliguria develops in about 50% of cases, particularly in older patients.
NSAID-associated Interstitial Nephritis
- Most frequent and clinically important
- Can occur w/over-the-counter agents like ibuprofen
- Devo of AIN more common in older populations
- Nephrotic range proteinuria may occur, but hematuria is rare
- Histologically pure lesion w/w/o papillary necrosis w/no glomerular disease (minimal proteinuria)
- Mix variety w/minimal change glomerular nephrosis also seen histologically- nephrotic range proteinuria and AIN
- Long term excessive use can result in chronic TIN
What other processes are associated with AIN?
- Bacterial infections: Corynebacterium diphtheriae, Legionella, E. coli, Staphylococci, Streptococci, Yersinia, Brucella
- Viral infections: CMV, EBV, Hantaviruses, Hep C, Hep B, HSV, HIV, Mumps, Polyoma virus, Measles
- Other infections: Leptospira,Mycobacterium, Mycoplasma, Rickettsia, Syphilis, Toxoplasmosis
- Immune and Neoplastic disorders: acute rejection of a renal transplant, SLE, Sarcoidosis, Glomerulo-nephritis, Lymphoproliferative disorders, Necrotizing vasculitis, Plasma cell dyscrasias, Sjogren’s Syndrome
- Primary or idiopathic AIN
- TINU syndrome: AIN and Uveitis
Aminoglycoside nephrotoxicity

- Recognized potential for causing acute renal failure in hospitalized pts (use has dramatically declined)
- Drug enters tubular lumen by glomerular filtration, is reabsorbed by proximal tubules, & tubule cell injury leading to necrosis may occur
1. 5% administered dose retained in epi cells lining S1, S2 segments of prox tubules, mainly in endosomal, lysosomal vacuoles, some Golgi
2. In cytosol, drug associates w/various organelles, like mito membranes and nuclei, which can initiate additional cascade of events leading to renal proximal tubule injury
3. Endocytosed by tubular cells -> much higher IC concentration than plasma concentration; tubular uptake mech has saturation kinetics - Manifested clinically by progressive INC in serum creatinine, renal K+, Mg++ wasting, renal glucosuria
- A lot of antimicrobial killing effect for several hours after its gone (once a day dosing)
- Can also injure the thick ascending limb, and look like Barterr’s syndrome
What are the risk factors for aminoglycoside nephrotoxicity?
- High or repeated doses or prolonged therapy
- CKD, diabetes
- Volume depletion
- Advanced age
- Renal ischemia or other nephrotoxins
What is contrast-induced nephropathy?
-
Renal tubular ischemia: short-term (~30 min) INC in renal blood flow (RBF), then prolonged renal artery vasoconstriction + DEC RBF + shunts residual BF from underperfused medullary to cortical segments
1. Direct tubular toxin - Serum creatinine: usually INC w/in 48-72 hours after contrast medium admin, reaches peak at 3–5 days, then returns to baseline within 7–10 days
- Urinalysis: renal tubular epi cells and coarse granular casts (not specific, but always abnormal)
- Non-oliguric: won’t know this pt has acute renal failure until you MEASURE SERUM CREATININE
- Required for arteriograms, angiograms, other diagnostic testing
- Doesn’t typically cause damage, but more likely in ppl with underlying DEC GFR (diabetics, CKD, etc.)
- No proteinuria or pyuria
What are the risk factors for contrast-induced nephropathy?
-
Established risk factors:
1. Preexisting renal insufficiency
2. Diabetes mellitus: + normal renal function = fairly low risk; + preexisting renal insufficiency = extremely high risk
3. Volume of contrast
4. Intravascular volume depletion -
Possible risk factors:
1. Congestive heart failure
2. Recurrent contrast procedures
3. Multiple myeloma
What do you see here? What are the causes?

- Papillary necrosis
- Papillae (inner medulla) susceptible to ischemic injury bc low blood flow (only about 10% of renal flow)
- Renal papilla: location where renal pyramids in the medulla empty urine into minor calyx in the kidney; histologically marked by medullary collecting ducts converging to form papillary duct to channel the fluid

What is analgesic abuse nephropathy?
- Initial occurrence reported in assoc w/phenacetin abuse -> textile industry workers took analgesics to relieve headaches
- Associated w/long-term use of analgesic mixtures containing phenacetin (acetaminophen) and aspirin or other NSAIDs
- Drug accumulates and is highly concentrated in the renal medullary interstitium

What are the clinical features of analgesic abuse nephropathy?
- Slow, progressive impairment of renal function
- Tubular dysfunction: devo of hyperkalemic, hyper-chloremic renal tubular acidosis and nephrogenic diabetes insipidus
- Impairment of sodium reabsorption
- May progress to the devo of papillary necrosis
- Uro-epithelial cancer (high frequency)
What is aristolochic nephropathy?
-
Chronic interstitial nephritis:
1. Principally in the cortex
2. Extensive interstitial fibrosis & tubular atrophy (end stages of these interstitial diseases -> bad)
3. Cellular infiltration of the interstitium scarce
4. Glomeruli are relatively spared and immune deposits are not observed - Findings suggest 1o lesions may be centered in the vessel walls, leading to ischemia & interstitial fibrosis
- Mechanism: drug forms covalent adducts with DNA
What are the clinical features of aristolochic nephropathy?
- Slowly progressive renal insufficiency
- Unremarkable urine sediment (bland)
- Waxy casts: form in dilated, atrophic tubules
- Proteinuria usually < 1gram/24 hour
- Extremely high incidence of cellular atypia and urothelial (transitional cell) carcinoma of renal pelvis, ureter, and bladder
- Ingestion of aristolochic acid has also been ID’d as cause of Balkan (familial interstitial) nephropathy
What are the clinical features of Balkan endemic nephropathy?
- Slowly progressive renal insufficiency
- Urine sediment usually unremarkable
- Proteinuria usually <1.0 g/day
- Renal tubular dysfunction
- Hypertension in <25% of patients
- Gross hematuria: may be a sign of uroepithelial tumor
What is Chinese herbs nephropathy?
- Rapidly progressive interstitial nephropathy attributed to weight-reducing diets containing Chinese herbs
- Renal pathology closely resembles characteristic lesions of Balkan endemic nephropathy
- Multiple foci of cellular atypia in renal pelvis, ureters
- Aristolochic acid, a known carcinogen and suspected etiologic agent
What is this?

Acute interstitial nephritis
What is gadolinium?
- Thought they could use this instead of iodinated contrast, but causes horrible skin lesions + interstitial fibrosis of the kidney (nephrogenic systemic fibrosis)
- Can’t give this anymore to someone with estimated GFR below 30
- Changed the product, so not in the same form
What is urate nephropathy?

What are the causes and mechanism of nephrocalcinosis?
-
Hypercalcemia: may induce formation of Ca stones and depo of calcium in kidney (nephrocalcinosis)
1. Hyperparathyroidism, multiple myeloma, Vit D intoxication, metastatic cancer, excess Ca intake (milk-alkali syndrome) - Earliest damage to tubular epi cells in form of mito distortion and cell injury, then Ca deposits in mito, cyto, and BM -> calcified cellular debris may obstruct tubular lumens, causing obstructive nephron atrophy and secondary interstitial fibrosis and inflammation
-
Earliest functional defect is inability to concentrate urine, but tubular acidosis and salt-losing nephritis, may also occur
1. With further damage, slowly progressive renal insufficiency develops
How does the kidney attempt to correct nephrocalcinosis?
- Ca sensor at thick ascending limb down-regulates NK2C transporter, reducing Ca reabsorption, but this makes you more apt to have kidney stones because you have more calcium making it to CD -> another Ca sensor in CD down-regulates aquaporins, “flushing out” the calcium with water
1. The more water you have, the more calcium you can dissolve in it - Calcium also constricts vessels, and can lead to intense vasoconstriction
What is acute phosphate nephropathy?
- Extensive accumulations of Ca-phosphate crystals in tubules can occur in pts consuming high doses of oral phosphate in preparation for colonoscopy
- Pts not hypercalcemic, but excess phosphate load, perhaps complicated by dehydration, causes marked precipitation of Ca-phosphate, typically presenting as renal insufficiency several weeks after exposure
- Pts typically only partially recover renal function
- These colon preps are now off the market
- CKD: will absorb phosphate normally, but not be able to excrete it
What is polycystic kidney disease?
- Auto dom: most common genetic kidney disease (5 -10% ESRD pts)
1. Spontaneous muts also possible - High penetrance, and leads to progressive CKD in adulthood, usually > age 40
- Cysts may occur in other organs, esp. liver
- Assoc w/intra-cranial aneurysms (vascular changes)
- Cysts can hemorrhage (gross hematuria, flank pain) and become infected
- Not associated with higher risk of renal cancer
- ADPKD1 and ADPKD2
What do you see here?
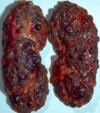
- Polycystic kidney disease: ID’d via imaging studies, ultrasound or CT iamging
- Cysts develop early, 20 -30 yr of age: progressive INC in # and size of cysts, kidney size/vol increasing
1. Normal anatomy becomes distorted, leading to CKD and ultimately ESRD - Hypertension is common
- Bland urinalysis, unless having hematuria (tubular pattern urinalysis, so minimal proteinuria)
What genes are associated with PCKD?
- Mutation of polycystin 1 or polycystin 2
1. PKD1: chrom 16 (80-90% of pts)
2. PKD2: chrom 4 - Genes encoding proteins w/in 1o cilia of renal tubular cells; likely involved w/cell-cell, cell-matrix interactions -> can be thought of as a ciliopathy
- For sibling to donate, has to be proven they don’t have it (i.e., also over 40 and don’t have cysts yet)
What is multiple myeloma?
- Neoplastic (malignant) proliferation of a single clone of plasma cells in bone marrow
-
Major laboratory diagnostic criteria:
1. >10% plasma cells in bone marrow
2. Complete or incomplete monoclonal Igs in serum and/or urine at elevated concentrations -
Monoclonal Immunoglobulins (antibodies):
1. Monoclonal, M, or paraproteins
2. Non-functional -> hypogammaglobulinemia
3. Light chain made in excess (freeely filtered at glomerulus)
4. Light chains reabsorbed by proximal tubule
What is myeloma kidney? Describe the pathogenesis, contributing factors, and prognosis.
-
Two main pathogenetic mechanisms:
1. Intra-tubular cast formation (distal)
2. Direct tubular toxicity by light chains (can get amyloid deposits in glomerulus -> proteinuria) -
Contributing factors to presence of renal failure due to multiple myeloma:
1. High rate of light chain excretion (tumor load)
2. Concurrent volume depletion - Prognosis: see attached chart
- Prox tubule cell endocytoses these and converts them to AA -> can overload this system, leading to FANCONI’S (generalized proximal tubule problems)

Where do MM light chains damage the renal tubules?
- Proximal tubule: direct tubular cytotoxicity
- Distal tubule: cast injury
- Most common injury you see myeloma cast injuries
1. Light chain tightly adhering to Tam-Horsfall protein (that forms backbone of all casts in urine), making myeloma casts, leading to tubular obstruction and injury -> MOST COMMON PRESENTATION FOR MYELOMA INVOLVING KIDNEY (large proportion of these pts)

What are the histologic features of myeloma kidney?

What is this?

Myeloma kidney
What do you see here?

- Myeloma kidney: note the myeloma casts and healthy glomerulus
If a UA comes back + for blood, but no RBC’s, what should you be thinking?
Myoglobin (look for muscle damage) or hemolysis
What should you be thinking if UA is negative for protein (urine albumin creatinine and urine protein creatinine ratios also low), but 24-hr urine has 3gm protein?
- UA is specific for albumin
- Suggests non-albumin protein in urine, i.e., light chains
What do you see here?
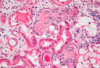
- Myeloma cast nephropathy -> myeloma kidney
- Many dilated tubules obstructed by densely eosinophilic hard casts, w/giant cell reaction and inflammatory infiltrates
- Also vacuolation and degeneration of tubules
What is the relationship between ATN and AKI? Between ischemia and ATN?
- ATN is a subset of AKI, the worst cases (% not really known because rarely biosied)
- Ischemic ATN is a subset of ATN (around 75%)
What do you see here? 3 characteristics?

- Gross pathology of acute tubular necrosis
-
Characteristics:
1. Enlarged - up to 30% over normal
2. Pale cortex
3. Congested medulla, esp. at cortico-medullary junction
When, how, and why does ATN lead to what you see here?

- Cortical hemorrhages
- When? Thromboemboli or septic emboli or some kind of uneven process
- How? Some areas get it worse (something involving blood vessels out near the cortical surface)
- Why? Septic shock
What are all of these things?
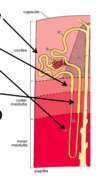
- Proximal convoluted tubule
- Proximal straight tubule
- Thin limb of loop of Henle
- Thick ascending limb of loop of Henle
- Distal convoluted tubule (8)
- Connecting segment (to collecting duct) (9)
- Collecting duct (10) (not part of the tubule)
The portions of the renal tubules most vulnerable to acute ischemic necrosis are? Why?
- Proximal straight tubule and thick ascending limb
- These are the two portions of the tubule in the outermost medulla at the corticomedullary junction, the area that gets congested with acute tubular necrosis -> vulnerable to hypoxia
What do you see here?

- Baseline normal renal tubules
- Cuboidal cells with granular, eosinophilic cytoplasm
What is this?

- Acute tubular necrosis
- Blebbing of luminal side of cell membranes, an early ischemic change -> reversible because glomeruli are still okay (tubules are much more susceptible to ischemia)
What is going on here?
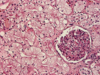
- Acute tubular necrosis
- Diffuse edema of tubular cells, and vacuolization of cytoplasm
- Early ischemic change (also seen with osmotic diuresis) -> reversible b/c glomeruli are still okay (tubules are much more susceptible to ischemia)
What do you see here?
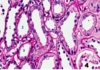
- Acute tubular necrosis
- Loss of brush borders, flattening of epithelium (evident in tubules 1,2,3)
- Necrosis & sloughing of epi cells (seen in tubule 4, lower right)
- PAS stain helps you see brush borders
What do you see here?

- Acute tubular necrosis
- Necrosis & sloughing of tubular cells mixing into casts with proteinaceous material
The apoptotic renal tubular epithelial cells are most likely have decreased cytoplasmic activity of?
BCL-2
What is going on here? What is the black arrow pointing at?
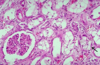
- ATN: vacuolization of the tubules in the top right
- Black arrow points to calcium oxalate from ethylene glycol
- Note: symptoms of ethylene glycol poisoning start with confusion, nausea, vomiting, appearance of intoxication, progress to hyperventilation and CHF, and then lead to acute kidney injury via the formation of calcium oxalate crystals -> lower back pain
How do treat hyperkalemia?
- Potassium over 7 mmol/L needs immediate tx
1. IV calcium gluconate: antagonizes membrane depolarization and protects against cardiac arrhythmias
2. IV insulin (+ glucose): drives potassium from bloodstream into cells - Potassium-binding resins (kayexalate): rectally or orally (don’t work immediately, but will help)
What do you see here?

- Gross pathology of acute pyelonephritis
- Dark red congestion and areas of light tan, suppurative inflammation -> some with necrosis, some coalescing into abscesses (upper right)
What do you see here? What is the arrow pointing to?

- Acute pyelonephritis: congestion with some mild hemorrhage (arrow)
- Heavy infiltration by leukocytes (blue dots, can’t tell they are polys at this low power)
What is this?

- Acute pyelonephritis: diffuse infiltration by leukocytes
- Can’t tell that they are neutrophils at this low power, but they are
Acute pyelonephritis is characteristically abscessing because it produces?
Liquefactive necrosis
What do you see here?

- Early r**e**nal abscesses
- Microscopy would show severe acute inflammation and necrosis, but not much liquefaction, although a couple in the cortex have dimples where the liquid has gone
- Learning this gross pathology will be most helpful for future radiologists -> they are the ones who will drain these
What is going on here?

- Microscopic renal abscesses
- So blue due to nuclear debris from breakdown of dead cells, esp. neutrophils
- Note the cracks -> later abscesses will have holes in them
What are some of the complications of acute pyelonephritis?
- Pyonephrosis
- Perinephric abscesses
- Healing by scar formation and deformation of calyx
- Acute papillary necrosis
What do you see here?

- Pyonephrosis and perinephric abscesses: possible complications of acute pyelonephritis
What do you see here?

- Cortical scarring: pyelonephritis can extend all the way out to the capsule, causing necrosis and then scarring visible on the cortical surface
Where does pyelonephritic infection come from?
- Can 1) ascend from the bladder (E. Coli) or 2) arrive via the bloodstream (staph aureus)
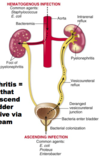
What do you see here?

- Gross pathology of chronic pyelonephritis: more likely from the ascending route due to the large size of the scars (and because “common things are common”)
- Scarring causes depression of the cortical surface
What is the relationship between AIN and drug reactions?
- AIN due to drug reactions a subset -> around 75%
- But, most drug reactions do not cause AIN
What do you see?

- Analgesic nephropathy
- Chronic interstitial nephritis and papillary necrosis
- Corticomedullary junction inflammation, liquefactive necrosis of medulla
What is this?

- Papillary necrosis (ischemic medulla) due to analgesic nephropathy (chronic interstitial nephritis)
The 51-year-old man with this CT scan has chronic renal failure due to?
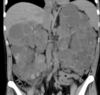
- Autosomal dominant (adult) polycystic kidney disease: slowly expanding cysts compress normal tissue, causing ischemic atrophy
- Mutations in polycystin-1 (85%) or polycystin-2 leading to ciliopathy, defective mechanosensing of urine flow, dysregulation of cell adhesion
- Two-hit genetics just like retinoblastoma
- Not usually removed, but can cause refractory HTN, and need to be removed
At what age does the average patient with auto dom PCKD need dialysis or transplantation?

- Age 50
- Frequently have intermittent gross hematuria
- HTN in 75% and is deleterious to the kidneys, as is UTI, so these are important to diagnose and treat
- Some of these patients also have cyst formation in the liver and up to 30% have cerebral vascular aneurysms that put them at risk for sudden death due to rupture and subarachnoid hemorrhage
What is going on here?

- Normal kidney on the left
- Auto dom (adult) PCKD kidneys on the right: cysts form at any level in the nephron, and gradually enlarge kidneys to huge sizes, up to 4kg per kidney
What is auto recessive PCKD?
- Child: numerous small cysts replace normal tissue during fetal life due to mutations in fibrocystin
- Often immediate, untreatable respiratory failure at birth due to pulmonary hypoplasia
Pause. Reflect.

Good job!
What is nephronophthisis?
- Medullary cystic disease complex: small kidneys with numerous small cysts at the corticomedullary junction and chronic tubulointerstitial nephritis and fibrosis
- Due to various mutations in 9+ genes for cilia components
- Most common genetic cause of end-stage renal disease (ESRD) in children
What is this?
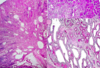
- Nephronophthisis: medullary cystic disease complex
- NOTE: kidneys will be small, rather than large like in PCKD
What do you see here?
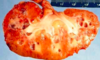
- Medullary sponge kidney: relatively common and usually innocuous condition
- Characterized by formation of cysts in the medulla, which generally do not impair renal function or affect the prognosis of the kidney or the patient as a whole
What is acute kidney injury?
- Abrupt impairment of renal function manifested by:
1. Increased creatinine
2. +/- oliguria
3. Acute tubular necrosis, it’s most common histopathologic counterpart
What is the most common cause of ATN?
- The most common cause of ATN is ischemia, and the most common cause of this ischemia is shock, especially septic shock
Why are the renal tubules of the outer medulla particularly susceptible to ischemic necrosis?
- Due to anatomic structural features required for the kidney to have the functional ability to concentrate urine
When will you see muddy brown granular casts?
- “Muddy brown” granular casts or tubular epithelial cell casts are a diagnostic feature of acute tubular necrosis in up to 80% of cases
What suggests the possibility of drug-induced AIN?
- New-onset azotemia with oliguria, fever, skin rash and especially eosinophilia
