Metabolic pathways and ATP production Flashcards
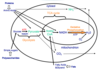
Metabolism overview: sketch a cartoon of the three stages of cellular metabolism that convert food to waste products, identify the cellular location of each stage;
Cellular Metabolism:
Glycolysis - Oxidation of glucose within the cytosol of individual cells, generating ATP and NADH.
TCA Cycle - Further oxidation of small molecules generated by glycolysis within the mitochondria of individual cells, generating ATP, NADH, FADH2 and waste products.
Oxidative Phosphorylation - generation of ATP within the mitochondria by the reduction of oxygen to water.
Glucose metabolism: explain the metabolism of glucose during glycolysis and recall the key reactions
Glycolysis overall: an anaerobic process, occurring in the cytoplasm of cells
1 x 6 carbon molecule (Glucose) ———–> 2 x three carbon molecules (pyruvate)
Yields 2 ATP
Substrate-level phosphorylation: the production of ATP by the direct transfer of a high-energy phosphate group from an intermediate substrate in a biochemical pathway to ADP
Within the ten reactions that make up the glycolysis pathway, there are two main concepts:
(i) Formation of a High Energy Compound -> involves the investment of energy in the form of ATP
(ii) Splitting of a High Energy Compound -> produces useful energy in the form of ATP generation
Step 1:
- Adding a phosphate is essentially irreversible
- Commits cell to glycolysis
- Traps glucose inside the cell
- enzyme; Hexokinase
- What we have: Glucose 6 phosphate
- What we’ve used: One ATP
Step 2 and 3:
- The isomerisation shuffles the glucose chair to give fructose
- The logic behind this reaction is that fructose can be split into equal halves when subsequently cleaved
- A highly symmetrical, high energy compound is generated
- enzyme;(for 2nd reaction) Phosphoglucose isomerase
- enzyme; ( for 3rd reaction) Phospofructose kinase
- What we have: Fructose 1,6 bisphosphate
- What we’ve used: 2 ATP
Step 4:
- Opening of the fructose ring to generate two high energy compounds
- enzyme; Aldolase
- What we have: 1 Glyceraldehyde-3-phosphate AND 1 Dihydroxyacetone phosphate
- What we’ve used: 2 ATP
Step 5:
- Enzyme used: Triose Phosphate Isomerse (TPI) (deficiency of TPI is severe metabolic disease)
- What we have: 2 Glyceraldehyde-3-phosphate
- What we’ve used: 2 ATP
Step 6:
- NADH is generated here which can be later used to generate yet more ATP within the mitochondria (oxidative phosphorylation )
- enzyme; Glyceraldehyde 3-phosphate dehydrogenase
- What we have: 2 1,3 bisphosphoglycerate
- What we’ve used: 2 ATP, 2 NAD 2 Phosphate groups
- What we’ve made: 2 NADH
Step 7:
- enzyme; Phosphoglycerate kinase
- What we have: 2x 3phosphoglycerate
- What we’ve used: 2 ADP, 2 NAD, 2 Phosphate groups
- What we’ve made: 2 NADH, 2 ATP
Step 8:
- enzyme; phosphoglycero-mutase
- What we have: 2 x 2phosphoglycerate
the rest is unchanged - What we’ve used: 2 ATP, 2 NAD, 2 Phosphate groups
- What we’ve made: 2 NADH, 2 ATP
Step 9:
- enzyme; Enolase
- What we have: 2 x phosphoenolpyruvate
- What we’ve made; 2x H2O
Step 10:
- Transfer of the high energy phosphate group to ADP, generating one ATP molecule in the process.
- enzyme; Pyrovate Kinase
- What we have: 2 x pyruvate
- What we’ve used: 2 ATP, 2 NAD, 2 Phosphate groups
- What we’ve made: 2 NADH, 4 ATP
recalling the key reactions, in particular those reactions that consume ATP and those that generate ATP (1, 3, 6, 7, 10 )

Recall and explain the fates of pyruvate
Pyruvate has Three Possible Fates
(i) Alcoholic Fermentation
This is characteristic of yeasts and can occur under anaerobic conditions.
(ii) Generation of Lactate
This is also anaerobic and is characteristic of mammalian muscle during intense activity when oxygen is a limiting factor.
Enzyme lactate dehydrogenase catalyzes this reaction
Lactate Dehydrogenase (LDH) as a Diagnostic Tool
- LDH is present in many body tissues, especially the heart, liver, kidney, skeletal muscle, brain blood cells and lungs
- Elevated levels can be used to diagnose: stroke, heart attack, liver disease (e.g. hepatitis), muscle injury, muscular dystrophy, pulmonary infarction
Creatine Phosphate
In muscle, the amount of ATP needed during exercise is only enough to sustain contraction for around one second.
Thankfully a large reservoir of creatine phosphate is on hand to buffer demands for phosphate
creatine phosphate – (creatine kinase) –> creatine+ATP
Creatine Kinase as a Diagnostic Tool
- When a muscle is damaged, creatine kinase leaks into the bloodstream.
- Elevated levels can be used to: diagnose myocardial infarction (heart attack), determine the extent of muscular disease, evaluate causes of chest pain, help discover carriers of muscular dystrophy (Duchenne)
Regeneration of NAD+ is essential
Both alcoholic fermentation and the generation of lactate serve one common purpose:
- They allow NAD+ to be regenerated and thus glycolysis to continue, in conditions of oxygen deprivation. i.e. conditions in which the rate of NADH formation by glycolysis is greater than its rate of oxidation by the respiratory chain.
- NAD+ is needed for the dehydrogenation of glyceraldehyde 3-phosphate, which is the first step in generating ATP for the body.
(iii) Generation of acetyl CoA
- This is the committed step for the entry of pyruvate into the TCA cycle and the overall reaction is as follows:
pyruvate + CoA + NAD+ –> acetyl CoA + CO2 + NADH
- This series of reactions occurs in the mitochondria of the cell and is catalyzed by The Pyruvate Dehydrogenase Complex
- The acetyl CoA thus formed is committed to entry into the citric acid cycle and can ultimately produce ATP by the process of oxidative phosphorylation
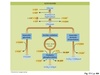
Explain the oxidative decarboxylation reaction catalyzed by pyruvate dehydrogenase, with reference to the product and the five co-enzymes required by this enzyme complex.
The Pyruvate Dehydrogenase Complex
is gigantic and consists of three individual enzymes and five co-factors:
- Thiamine pyrophosphate (TPP)
- Lipoamide
- FAD (Flavine Adenine Dinucleotide)
- CoA
- NAD+
Thiamine pyrophosphate (TPP)
- Derivative of vitamin B1 (thiamine)
- Readily loses a proton and the resulting carbanion attacks that of pyruvate to yield hydroxyethyl-TPP
- Deficiency of Vitamin B1 causes Beri-Beri - symptoms include damage to peripheral nervous system, weakness of musculature and decreased cardiac output.
Lipoamide
- Functional group (undergoes oxidation and reduction).
- Long arm allows dithiol group to swing from one active site to another.
- Arsenite (AsO33-) and mercury have a high affinity for neighbouring sulphydryl groups, such as those that occur in reduced lipoamide and will readily inhibit pyruvate dehydrogenase.
Flavine Adenine Dinucleotide (FAD)
FAD accepts and donates 2 electrons with 2 protons (2 H):
FAD + 2 e- + 2 H+ FADH2
Some co-factors are permanently bound to enzymes:
- Enzyme: pyruvate decarboxylase
Prosthetic Group: thiamine pyrophosphate (TPP) - Enzyme: lipoamide reductase-transacetylase
Prosthetic Group: lipoamide - Enzyme: dihydrolipoyl dehydrogenase
Prosthetic Group: FAD (Flavine Adenine Dinucleotide)
How the complex works (image):
- Decarboxylation of pyruvate to give hydroxyethyl TPP.
- Oxidation & transfer to lipoamide to give acetylipoamide.
- Transfer of the acetyl group to CoA to give acetyl CoA.
- Regeneration of oxidised lipoamide.
- Regeneration of oxidised FAD, generating NADH.
Distinguish between the aerobic and anaerobic metabolism of glucose and outline what happens in cancer
Aerobic Respiration
- 38 ATP
- Continues past glycolysis
- Enters TCA cycle and electron transport system
Anaerobic Respiration
- 4 ATP
- Stops at glycolysis
- Pyruvate converted to lactate
- Lactate processed in liver
Cancer Glucose Metabolism
- Glucose is rapidly metabolized to lactate by aggressive tumor cells
- We can make radiolabelled glucose – this will go the cells using a lot of glucose
- PET scan detects the location of the radiolabelled glucose
- Cancer cells undergo selective reprogramming and use several mechanisms to increase the flux of glucose in glycolysis to aid tumour growth:
Increased expression of glucose transporters (e.g. GLUT1)
Increased expression of hexokinase and phosphofructokinase.
The tricarboxylic acid (TCA) (Krebs) cycle: summarise the processes by which glucose, fatty acids and amino acids lead to products that can enter the TCA cycle, and explain the oxidation of acetyl-CoA with the formation of NADH and FADH2 by the TCA cycle
Pyruvate can be used to make Acetyl CoA
pyruvate + CoA + NAD+ –> acetyl CoA + CO2 + NADH
- catalyzed by pyruvate dehydrogenase complex
- making NADH from NAD+
These reactions occurs in the mitochondria of the cell.
The acetyl CoA thus formed is committed to entry into the citric acid cycle.
The Krebs Cycle (The Tricarboxylic Acid (TCA) cycle or The Citric Acid Cycle)
A continuous cycle of eight reactions, starting with 2 carbon atoms from acetyl CoA being condensed with the 4 carbon unit of oxaloacetate to give a 6 carbon unit, citrate.
Rn #1 Transfer to the oxaloacetate of 2C from acetyl CoA.
Rn #2 Isomerisation of citrate to give isocitrate.
Rn #3 Oxidation of isocitrate to give a-ketoglutarate.
Rn #4 Similar to the reaction catalysed by PDH (lecture 3).
Rn #5 CoA is displaced by a phosphate molecule which is subsequently transferred to GDP to form GTP.
GTP = Guanosine Triphosphate
- Guanosine Triphosphate (GTP) itself can act as a regulator of small GTP binding proteins or play a role in signal transduction processes
- An alternative isoform of succinyl CoA synthetase (A-SCS) is found in skeletal and cardiac muscle and catalyses the same reaction but generates ATP from ADP
Rn #6 Oxidation of succinate generating some FADH2.
Rn #7 Addition of a water molecule, breaking a double bond.
Rn #8 The last step. Dehydrogenation of malate to give oxaloacetate, the starting point of the cycle.
Pneumonic:
- Can - citrate
- I - isocitrate
- Keep - a ketoglutarate
- Selling - succinyl coA
- Sex - succinate
- For - fumarate
- Money -malate
- Officer? -oxaloacetate
(need to know 6 out of the 8 reactions.The 2 you technically don’t need to know is: citrate -> isocitrate and Fumarate -> malate)
- Each turn of the cycle produces two molecules of CO2 (waste) plus three molecules of NADH, one molecule of GTP and one molecule of FADH2.
- TCA: 8 steps, involving 8 enzymes
- All enzymes are in the mitochondrial matrix
- Exception: Succinate dehydrogenase, on inner surface of mitochondrial membrane
- Operates under aerobic conditions, as the NAD+ and FAD needed are only regenerated via the transfer of electrons to O2 during oxidative phosphorylation
Glycolysis and TCA Cycle Provide the Starting Point for Many Biosynthetic Reactions (Metabolites can leave the pathway and be used as building blocks for more complex molecules)
TCA defects in Cancer
- Some enzymes can be mutated to reduce TCA activity
- Cancer cells depends on anaerobic respiration for ATP
- This still happens in high oxygen settings – the Warburg Effect
State the products of glycolysis, the link reaction, and TCA cycle by recalling their steps.
What has been made so far:
1 glycose –> 2 ATP + 10 x NADH + 2 x FADH2 + 2 GTP
10 x NADH: Makes 30 ATP overall (since One NADH makes 3 ATP)
2 x FADH2: Makes 4 ATP overall (since One FADH2 makes 2 ATP)
By stage:
Glycolisis: 2 ATP, 2 NADH
Link reaction: 2 NADH
TCA cycle: 6 NADH, 2 FADH2, 2 GTP
Outline the use and significance of transamination reactions in protein metabolism
- Loads of things can feed into the Kreb’s Cycle: proteins included
- The general strategy of amino acid degradation is to remove the amino group (which is eventually excreted as urea) whilst the carbon skeleton is either funneled into the production of glucose or fed into the Krebs cycle
- Degradation of all twenty amino acids gives rise to only seven molecules, pyruvate, acetyl CoA, acetoacetyl CoA, a-ketoglutarate, succinyl CoA, fumarate and oxaloacetate
Protein metabolism involves transamination reactions
Definition: a reaction in which an amine group is transferred from one amino acid to a keto acid thereby forming a new pair of amino and keto acids.
example: (image)
Alanine (C3) undergoes transamination by the action of the enzyme alanine aminotransferase.
alanine + a-ketoglutarate -> pyruvate + glutamate
Alanine is converted to pyruvate, that enters the Kreb Cycle
Glutamate is converted to a-ketoglutarate by glutamate dehydrogenase,
The amino group is removed
Amino group converted to urea and excreted from body
ALT = alanine aminotransferase –> Increased levels in the blood can indicate problems with the liver (ALT normally should be a lot low levels in blood)

Shuttle mechanisms: summarise the glycerol phosphate shuttle and state why it is required
NADH Transportation:
- NADH produced in glycolysis needs to enter the mitochondria to be utilized by the process of oxidative phosphorylation and to regenerate NAD+.
- Remember, there is only a finite amount of NAD+ and unless it is regenerated, glycolysis will very quickly grind to a halt.
- How does NADH cross from the cytosol into the matrix of the mitochondria
- The Glycerol Phosphate Shuttle – skeletal muscle, brain
- The Malate-Aspartate Shuttle - liver, kidney and heart
The Glycerol Phosphate Shuttle
Electrons from NADH, rather than NADH itself are carried across the mitochondrial membrane via a shuttle.
- Cytosolic glycerol 3-phosphate dehydrogenase transfers electrons from NADH to DHAP to generate glycerol 3-phosphate.
- A membrane bound form of the same enzyme transfers the electrons to FAD. These then get passed to co-enzyme Q, part of the electron transport chain (lecture 5).
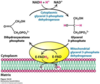
Shuttle mechanisms: summarise the malate-aspartate shuttle and state why it is required
The Malate-Aspartate Shuttle
This system uses two membrane carriers and four enzymes.
The net reaction in terms of NADH is:
NADH cytoplasmic + NAD+mitochondrial —>
NAD + cytoplasmic + NADH mitochondrial
- Transamination reactions occurs throughout – where an amine group is exchanged for a keto group between 2 molecules
- A hydride ion (H-) is transferred from cytoplasmic NADH to oxaloacetate to give malate, a reaction catalysed by cytosolic malate dehydrogenase (MDH).
- Malate can be transported across the membrane
- Malate is rapidly re-oxidised by NAD+ to give oxaloacetate and NADH ( catalysed by mitochondrial MDH)
- The other parts of the reaction is there to replenish oxaloacetate in the cytoplasm
image
A - alpha-ketoglutarate transporter, exchanges alpha-ketoglutarate for malate.
B - glutamate/aspartate transporter, exchanges glutamate for aspartate.

NADP
NADPH takes part in anabolic reactions, whereas NADH takes place in catabolic reactions.
The use of different co-factors for sets of reactions allows electron transport in catabolism to be kept separate to that of anabolism.
NADP+ is also an Electron Carrier
Like NAD+, NADP+ can pick up two high energy electrons and in the process, a proton (H+) collectively known as a hydride ion (H-).
NADP+ – + H+ + 2e—> NADPH
NAD+ –+ H+ + 2e—> NADH
The phosphate group of NADP+ does not participate in electron transfer, but gives it a slightly different conformation, meaning that it will bind to different enzymes than NAD+.
The hydride ion is held in a high-energy linkage, allowing it to be easily transferred to other molecules.
NADPH Functions:
- co-factor in the pathway involved in thymidine synthesis
- co-factor in the biosynthesis of Cholesterol
NADPH helps to catalyse the final reaction of several, that lead to cholesterol synthesis.
The C=C bond is reduced by the transfer of a hydride ion (two electrons plus a proton from solution, H-).
Mitochondria: Structure, Function and Evolution
The mitochondria
- Kreb’s Cycle happens in matrix
- Oxidative phosphorylation happens on inner membrane
- Folds of inner membrane (cristae) increase surface area
- Mitochondria can move in cells to areas of high demand
Mitochondria Evolution
- Mitochondria are prokaryotes that had a co-dependent relationship with eukaryotic cells
- Eventually, they lived in eukaryotic cells
Supporting evidence
- Mitochondria can only arise from pre-existing mitochondria and chloroplasts.
- Mitochondria possess their own genome and it resembles that of prokaryotes, being a single circular molecule of DNA, with no associated histones.
- Mitochondria have their own protein-synthesizing machinery, which again resembles that of prokaryotes not that of eukaryotes.
- The first amino acid of their transcripts is always fMet as it is in bacteria and not methionine (Met) that is the first amino acid in eukaryotic proteins).
- A number of antibiotics (e.g., streptomycin) that act by blocking protein synthesis in bacteria also block protein synthesis within mitochondria and chloroplasts. They do not interfere with protein synthesis in the cytoplasm of the eukaryotes.

Electron transport and oxidative phosphorylation: summarise the electron transport chain in mitochondria, explain the chemiosmotic model, and explain how ATP synthase is able to either use or generate ATP with reference to its structure; explain the mechanisms of action of metabolic poisons
What is oxidative phosphorylation?
- Using energy from NADH and FADH2 to make ATP
- Electrons are released from NADH
- Electron transport chain uses these high energy electrons to make ATP
The Electron Transport Chain
Enzymes:
- NADH Dehydrogenase complex
- Cytochrome b-c1 complex
- Cytochrome oxidase complex
Carriers:
- Ubiquinone (a.k.a. co-enzyme Q)
- Cytochrome C.
- These proteins accept electrons and in doing so, a proton (H+) from the aqueous solution. As electrons pass through each of the complexes, a proton is passed or ‘pumped’ to the intermembrane space.
- Electrons move along chain, and lose energy
- each protein has a higher affinity for electrons as we go along
- The energy lost from the electrons is used to move protons into the intermembrane space
- At the end, electrons are combined with protons and oxygen to make water
- The free energy of the electrons decrease along the electron transport chain
- The Redox potential also becomes more positive (more likely to accept electrons)
Chemiosmotic Theory
2 steps to oxidative phosphorylation
- Movement of protons from mitochondrial matrix to inter membrane space
- Pumped protons are allowed back into the matrix through ATP synthase
This drives ATP production
Why do Protons want to go back to the matrix?
- Concentration gradient
- Transmembrane electrical potential
ATP synthase:
- is a multimeric enzyme consisting of a membrane bound part (F0: consists of a, b, c subunits) and a F1(:consists of alpha, beta and gamma parts) which projects into the matrix space.
- Protons can only go through ATP synthase
Mechanism:
- Proton flows through it
- C subunit rotates
- Attached gamma subunit also rotates
- Alpha and beta are stationary
- Fixed to cell membrane by B subunit
- The movement between the two makes ATP
Rotation of the enzyme drives transitions states, with altering affinities for ATP and ADP.
- As a consequence, conformational energy flows from the catalytic subunit into the bound ADP and Pi to promote the formation of ATP (chemical energy).
- The direction of proton flow dictates ATP Synthesis v ATP Hydrolysis –> depending on the direction of the flow of protons through the ATP synthase, the complex can either generate ATP or consume it.
- If protons go into the inter-membrane space, ATP synthase reverses in direction and breaks down ATP
Distinguish between a fatty acid and a triglyceride; distinguish between lipid catabolism and lipid anabolism
Fatty acid= basically just a hydrocarbon chain with –COOH on the end
This refers to if the acid is saturated with hydrogen or not: saturated= no more hydrogens can bind, so all single bonds
Triglyceride (=triacylglycerol, TAG)= one glycerol molecule with three fatty acid chains bound via ester bondsLipid metabolism:
- Lipid catabolism: Breaking fats down to make acetyl CoA (β oxidation)
- Lipid anabolism: Using acetyl CoA to build up fats (lipogenesis)
Summarise the pathways for the catabolism of fatty acids with respect to the substrates and products, coenzymes used, carrier molecules and their cellular locations
Breaking fats down to make acetyl CoA (β oxidation)
- acetyl CoA can be made by sugars but also fats
- why would we use fats?
- By weight, fat provides DOUBLE the ‘caloric yield’
- 50% of the body’s energy comes from fat EXCLUDING the brain
- What can the brain use? Glucose & Ketone bodies
Making acetyl CoA from a fatty acid
- Make an acyl CoA molecule out of the fatty acid
- Transfer this molecule inside the mitochondria via the carnitine shuttle
- Beta oxidation
1. Making an acyl CoA
- Fatty acids converted to acyl CoA before they can undergo beta oxidation
- The enzyme for this reaction is called an acyl coA synthetase
- You need 2 high energy bonds
Fatty acid + A spare CoA + ATP –acyl coA synthetase –> acyl CoA + AMP
(Basically just involved adding a CoA molecule onto the fatty acid, which requires 2 high energy bonds, so ATP -> AMP + PPi)
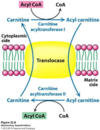
2. Getting the acyl CoA into the mitochondria using the carnitine shuttle (image)
- The acyl CoA cannot get through the inner mitochondrial membrane into the matrix.
- That’s why a shuttle is needed
- First, carnitine takes the acyl group from the acyl CoA, giving acyl carnitine. The CoA is left on its own
- The acyl carnitine can move through the membrane using a translocase
- Now, on the other side of the membrane, CoA picks up its acyl group from carnitine. Carnitine is now on its own, and can go back through the membrane using a translocase
- Beta oxidation of the fatty acid
- Finally we’re inside the mitochondrial matrix, which is where all the enzymes for beta oxidation are
- 4 reactions happen to the fatty acid molecule during beta oxidation, to split the fatty acid into acetyl CoA molecules
- Oxidation
- Hydration
- Oxidation
- Thiolysis
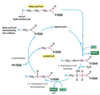
Explain the process of B oxidation
- Beta oxidation of the fatty acid
4 reactions happen to the fatty acid molecule during beta oxidation, to split the fatty acid into acetyl CoA molecules
- Oxidation
- Hydration
- Oxidation
- Thiolysis
Steps: (Think of hydrogens as electrons)
- Oxidation is happening because FAD receives a hydrogen molecule from the fatty acyl CoA
- Hydration is happening as you can see water being added to the oxidised fatty acyl CoA
- The second oxidation happens as another hydrogen is transferred from the acyl CoA molecule to the NAD+ (which itself is reduced)
- Thiolysis is happening as the acyl CoA molecule is split into an acetyl CoA molecule, and left behind is an acyl CoA molecule shortened by 2 carbons
* The cycle of beta oxidation will continue until the fatty acid is completely broken down
We often use palmitic acid when looking at beta oxidation
- It’s commonly found in plants and animals
- It has 16 carbons
- How many times does it need to undergo beta oxidation to be completely broken down into acetyl CoA molecules - 7
Overall reaction
What did we put in?
An acyl CoA (16C) so palmitoyl CoA (from palmitic acid)
7 FAD (From the first oxidation- step 1)
7 H2O (From the hydration- step 2)
7 NAD+ (From the second oxidation- step 3)
7 HS- CoA (These are needed to bind to the 2C group removed at the thiolysis stage of each cycle)
What did we get out?
8 acetyl CoA
7 FADH2 (FAD that we used to oxidise the acyl CoA is itself reduced)
7 NADH (NAD+ that we used to oxidise the acyl CoA is itself reduced)
Overall reaction of B oxidation:
Palmitoyl A + 7 FAD + 7 NAD+ + 7 H2O + 7 CoA —> 8 acetyl coA + 7 FADH2 + 7 NADH
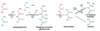
Explain the reasons and mechanisms of ketogenesis.
AFTER this process (using fats to make acetyl CoA to make energy), we have acetyl coA
- We need oxaloacetate (OAA) for acetyl CoA to enter the TCA cycle
- Sometimes there is not enough OAA, when?
- During fasting, fat is burnt, lots of acetyl CoA is made, and there is extra acetyl CoA compared to the OAA supplies, thus when fat metabolism exceed carbohydrate metabolism
- When there is not enough OAA, acetyl CoA can’t be used in Kreb’s
- acetyl CoA from fatty acids are used to make ketones
Ketone formation from fatty acids: image
Explain how inborn errors of fatty acid metabolism may cause disease
Diseases relating to fat metabolism
- Medium Chain Acyl CoA Dehydrogenase Deficiency (photo)
- Predominantly occurring in Caucasians.
- Occurs 1 in 10,000 live births in the UK per year.
- A family of different Acyl-CoA-dehydrogenases catalyze the initial step in each cycle of fatty acid β -oxidation within the mitochondria matrix.
Each Acyl-CoA-dehydrogenase can bind a fatty acid chain of varying lengths:
- Short-chain acyl-Co enzyme A dehydrogenase (<6C)
- Medium-chain acyl-Co enzyme A dehydrogenase (C6-C12)
- Long-chain 3-hydroxyacyl-Co enzyme A dehydrogenase (C13-C21)
- Very long-chain acyl-Co enzyme A dehydrogenase (>C22)
No cure for MCAD as of yet, so how do we treat?
- You can’t let them have a need to burn fat, i.e. they have to have enough glucose and cannot fast for more than 10 hours
- It’s a genetic disease: autosomal recessive
- 1/100 causes of Sudden Infant Death Syndrome (SIDS)
- The presenting patient would have needed iv. glucose
- Primary Carnitine deficiency
- Autosomal recessive disorder.
- Symptoms appear during infancy or early childhood and include encephalopathies, (cardiomyopathies, muscle weakness; and hypoglycaemia).
- Mutations in a gene known as SLC22A5 which encodes a carnitine transporter result in reduced ability of cells to take up carnitine, needed for the β-oxidation of fatty acids.
- Give a supplement –> ‘carnitor’
Summarise the pathways for the anabolism of fatty acids with respect to the substrates and products, coenzymes used, carrier molecules and their cellular locations
USING ACETYL COA TO MAKE FATTY ACIDS
Making fatty acids from acetyl CoA (lipogenesis)
Lipogenesis:
- Opposite of beta oxidation
(What were the four reactions in beta oxidation Oxidation, hydration, oxidation, thiolysis)
- The four reactions in lipogenesis are:
- DECARBOXYLATIVE CONDENSATION
- REDUCTION
- DEHYDRATION
- REDUCTION
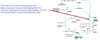
1.Decarboxylative condensation
Step 1: make the donator, malonyl CoA
We have acetyl CoA and using acetyl CoA carboxylase, we add a carbon (Bicarbonate is the source of carbon here)
Acetyl CoA + HCO3- +ATP –> Malonyl CoA + ADP + Pi
donator malonyl coA gives carbons to acetyl coA ONLY IF it is bound to APC (acyl carrier protein)
Step 2: Binding malonyl CoA to ACP
Malonyl coA + ACP –> Malonyl ACP + CoA
Step 3. Making acetyl ACP
Acetyl CoA + ACP –> Acetyl ACP + CoA
How many carbon molecules does malonyl have? 3
Step 4. Condensation of acetyl CoA and malonyl CoA
(image left)
So there is condensation of two carbon chains
Since malonyl has 3C –> it only actually donates 2 carbons, and the final carbon is lost as CO2 (HENCE it is decarboxylative)
Then (image right)
- reduction
- dehydration
- reduction
We made a fatty acid out of acetyl CoA:
- This process added 2 carbons to an acetyl CoA
- How many carbons does the new carbon chain have? -4
- What if the fatty acid I wanted to make palmitic acid?
Palmitic acid = 16C so repeat steps 4-7 another 6 times to get palmitoyl ACP - hydrolize palmitoyl ACP –> palmitic acid + ACP
Overall: 2 important enzymes
- Acetyl CoA carboxylase: catalyses carboxylation (step 1): acetyl CoA –> malonyl CoA
- Fatty acid synthase: big enzyme made up of seven smaller enzymes which catalyze all the reactions after the first one
Overall equation for lipogenesis:
What did we put in
- 1 acetyl CoA
- 7 malonyl CoA
- 14 NADPH
- 14 H+
What did we get out
- Palmitic acid
- 14 NADP+
- 8 HS-CoA
- 6 H2O
- 7CO2
Acetyl CoA (C2) + 7 Malonyl CoA (C3) + 14NADPH + 14H+ —-> Palmitate(C16) + 7CO2 + 6H2O + 8 CoA-SH + 14NADP+

Comparing beta oxidation and lipogenesis

Cholesterol: summarise the synthesis of cholesterol from acetyl-CoA
(up to now we have seen how from acetyl coA we have made ketones and fatty acids)
How cholesterol is made from acetyl CoA
OVERALL MECHANISM:
(in cytoplasm)
- Make IPP (isopentenyl pyrophosphate)
- Condense (i.e. join together) 6 IPP, making squalene
(in endoplasmic reticulum)
- Modification of squalene by monooxygenases to make cholesterol
1. Make IPP
step 1: put 2 acetyl CoA (2C) groups together
step 2: Add another acetyl CoA to that acetoacetyl CoA (so you’ve basically just added 3 acetyl CoAs together = 6C)
step 3: Reduce HMG-CoA (6C) to make mevalonate(6C)
HMG CoA reductase is important because:
- Negative feedback happens (from mevalonate and cholesterol/bile salts)
- Target of statins
step 4: Mevalonate undergoes:
- Sequential phosphorylation (position 3 and 5)
- Decarboxylation
IPP (5C) is made
2. Combine 6 IPPs
step 1. IPP + dimethylallyl pyrophosphate* (mi matheis onoma) –> Geranyl pyrophosphate (10C)
step 2. Geranyl pyrophosphate + IPP –> Farnesyl pyrophosphate (15C)
step 3. Farnesyl pyrophosphate + farnesyl pyrophosphate –> Squalene (+ 2PPi) (30C)
3) Squalene cyclized to cholesterol
Squalene (30C) —-> Lanosterol (30C) —– (19 steps & demethylation 3 methyl groups removed) –> cholesterol (C27)
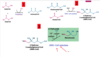
Explain the synthesis of bile acids, steroid hormones and vitamin D from cholesterol
Synthesis of bile salts
MAJOR BREAK DOWN PRODUCTS
Cholesterol —-> Glycocholate (=Primary bile salt)
Taurocholate
Bile
- salts are made in the liver and stored in the gall bladder
- After storage in gall bladder, released into small intestine
- Emulsify fats in the intestine, aiding their digestion and absorption of fats and also that of fat-soluble vitamins e.g. A, D, E and K.
Needed for digestion of?
- Fats - A lack of bile salts results the majority of fat passing through the gut undigested and unabsorbed resulting in steatorrhea (fatty stool).
- Fat soluble vitamins
Bile salts are the major breakdown products of cholesterol and account for about half of the 800 mg of cholesterol made each day by the liver.
Synthesis of hormones
Cholesterol —desmolase–> Pregnenolone —> All other steroid hormones
Pregnenolone is a precursor to all steroid hormones
Synthesis of vitamin D
7-dehydrocholesterol –(UV light)–> Pre-vitamin D –> Vitamin D
- Vitamin D made into calcitriol, involved in calcium absorption
- Vitamin D deficiency in children? -Rickets
Cell signalling
1) LIPID RAFTS in plasma membrane… Lipid rafts are fluctuating assemblies of cholesterol and sphingolipids, within a plasma membrane
They organize processes such as cellular signalling by localising key proteins such as cell surface receptors (light blue).
2) Cholesterol is also covalently attached to the N-terminal fragment of the hedgehog signalling protein (N-Hh) during its processing.
This limits its diffusion within tissues which is key to successful limb formation during embryogenesis.
Explain the mechanism of transport of cholesterol around the body and its uptake into cells, and the role of cholesterol in atherosclerosis
Absorption/transport of fats/cholesterol
a - Lipid digestion by lingual, gastric and pancreatic lipases results in the formation of Monoacylglycerol (MAG), diacylglycerol (DAG) and free FAs that are released by lipid hydrolysis and join with BS (Bile Salts), CL, (Cholesterol) lysophosphatidic acid (LPA) and fat-soluble vitamins to form mixed micelles.
b - Digested dietary products are absorbed by enterocytes that line the brush border of the small intestine. TAGs are resynthesized under the control of several enzymes prior to incorporation into chylomicrons (CM) and transportation via the lymphatics and on into the bloodstream.
Steps:
- In gut lumen, triglycerides from food have fatty acids removed, to make monoacylglyceride (MAG) or diacyglyceride (DAG) plus two or one free fatty acids by gut enzymes.
- Then, MAG and the free fatty acids are combined with bile salts and cholesterol to make a mixed micelle
- The mixed micelle can pass through cells lining the gut
- Now inside these cells, the MAG and fatty acids from the micelle are re-synthesised into triglycerides
- The triglyceride is then incorporated into a ‘chylomicron’, along with cholesterol and apoproteins. This then passes into the lympatic system via a lacteal
Chylomicrons are a type of lipoprotein (image)
- Chylomicrons in the bloodstream are detected by lipoprotein lipase enzyme on the inner lining of blood vessels
- This converts triglyceride back to glycerol and fatty acids
- The apoprotein of the chylomicron activates the lipoprotein lipase
- But there was also cholesterol in the chylomicron
- What happens to that?
- Leftovers of the chylomicron are taken up by the liver (including cholesterol)
- Liver incorporates cholesterol into a lipoprotein
LIPOPROTEINS:
- Lipoproteins are composed of a phopholipid monolayer containing cholesterol and proteins known as apoproteins such as Apo A-I, Apo B-100, or Apo E.
- Packed within the core of the lipoprotein are a mixture of cholesterol esters and triacylglycerols.
- Cholesterol esters are synthesized in the plasma from cholesterol and the acyl chain of phosphatidylcholine (lecithin) via a reaction catalyzed by lecithin:cholesterol acyltransferase (LCAT).
- This makes cholesterol esters more hydrophobic than cholesterol and allows them to pack more tightly within the lipoprotein core.
3 types to know about:
- CHYLOMICRONS
- LOW DENSITY LIPOPROTEINS:
BAD cholesterol.
Takes cholesterol from liver to peripheral tissues
- HIGH DENSITY LIPOPROTEINS
GOOD cholesterol.
Brings cholesterol back from periphery to liver, for use or disposal. REVERSE cholesterol transport
- Very low density lipoproteins (VLDL)
- Intermediate density lipoproteins (IDL)
Each type of lipoprotein has a varying apoprotein component which allows them to be recognized by different cell types.
LDL are HDL are principal lipoproteins and play key roles in cholesterol transport.
Familial Hypercholesterolaemia
- LDL is bad (takes cholesterol to periphery)
- LDL is taken up by the liver using the LDL receptor, by receptor mediated endocytosis
- People with familial hypercholesterolaemia have mutant/absent LDL receptor, so LDL left in circulation
- Dominant Trait
- They get atherosclerosis and heart attack
Drug treatment
Most people have hypercholesterolemia due to risk factors (obesity, diet, alcohol, diabetes), not mutation
Class of drugs:
- Statins: Inhibits HMG-CoA reductase, so limits cholesterol synthesis
- Resins: Prevents absorption of bile salt + cholesterol complex in small intestine

Recall elements of tissue specific metabolism
- Muscle (including heart) can utilise carbohydrate and fatty acid oxidation
- Brain and nervous tissue cannot use fatty acids, just glucose (and ketones)
- Adipose tissue stores fatty acids as triglycerides
- Liver stores carbohydrates glycogen
- Glycogen is formed from glucose-6-phosphate in which 2 tissues? 1) Liver 2)Muscle
- Happens in response to insulin release, as liver takes up more glucose, and stores it as glycogen
