Lecture #28 - Sequencing humans Flashcards
(21 cards)
- What’s DNA replication?
- What’s PCR?
- What’s recombinant DNA?
- What’s chain termination (Sanger) DNA sequencing?
- What’s alinging sequences?
- DNA double helix replicates itself by synthesising copies of each strand.
- Uses a pair of flanking primers to synthesise many copies of the region of DNA that lies in between.
- The universal code means bits of DNA can copied and stuck together with other bits
- Works by synthesis, making many labelled copies of the DNA and separating by size.
- Sequences of DNA that overlap can be aligned using computers and assembled into longer fragments or ‘contigs’.
DNA sequencing
- Based on what?
- What must be known already?
- What’s a primer?
- What’s the enzyme that copies DNA?
- What’s the bulding blocks of DNA?
- What’re dideoxyribonucleotides?
- It’s based on Fred Sanger’s “dideoxy method” - it’s for sequencing DNA that’s 800 to 1000 base pairs long
- A short length of sequence must be known already (around 20 bases). It can only elongate - not fully start from scartch.
- Primer = synthetic oligonucleotide complementary to the known sequence
- DNA polymerase = the enzyme which copies DNA (needs ATP, CTP, GTP etc)
- Bulding blocks of DNA = Deoxyribonucleotides
- . Dideoxyribonucleotides (flourestly tagged) are modified nucleotides which prevent further extension. Each dideo has a different flourest tag - each will be different signal when activated by laser

Many copies of DNA are made
Each primer is extended by…….until…..
What’s generated?
Each primer is extended by DNA polymerase until a dideoxynucleotide is incorporated.
Synthesis of each new strand starts at the 3’ end of the primer and continues until a ddNTP happens to be inserted instead of the equivalent dNTP

The DNA fragments are passed though…….
Sequence is read by……
Conclusion?
The DNA fragments are passes though a capillary to separte them by size
Sequence is read by a detector which sees the flourescent tags
Conclusion: The colour of the flourescent tag on each strand indicates the identity of the nucleotide at its 3’-end. The results can be printed out as a spectogram and the sequence, which is complementary to the template strand can then be read from the bottom (shortest strand). The sequence here begns after the primer

How do we sequence genomes today?
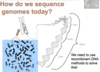
Step 1 of How we sequence now
Smash it up!
Break DNA from each chromosome into overlapping chunks (so can stick em back together)
Break up many copies of the chromosomal DNA with ultrasound waves (random)
This image shows three copies of the same chromosome

Step 2 of How we Sequence now
Add linkers
Ligate (tie up) pieces of known sequence to each end (linkers)
-Basically add the same set of linkers (for which you have made primers) to each DNA fragment and sequence each one

Step 3 of how we sequence now
Sequence using primers to the linkers
Use the same known linker as a primer to shotgun sequence all the unknown bits of DNA from the genome, using ‘massive parallel sequencing’ platforms
Picked up by high speed camera

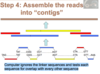
Step 4 of how we sequcne now
Assemble the reads into “contigs”
Computer ignores the linker sequences and tests each sequence for overlap with every other sequence
Puts em together into one chromosome based on overlap
You can put multiple ppl’s DNA into one machine bc barcodes differently.
Genome sequencing challenges?
3 points
- Mistakes can occur (as in DNA replication) so the entire genome is sequenced several times over. Information about variation is retained (we’ll come back to why this is in later lectures).
- Assembling the short sequences requires a huge amount of computing power.
- Confusing parts such as repetitive regions can be resolved by comparing to a “reference genome” or maps.
We thought there were intially like 35k genes but current data says only 20.5k - why?
- Size of the genome is not indicative of the number of genes
- Many genes turned out to be pseudogenes- defective non-functional copies of true genes (dont code for proteins and theyre kinda just decaying away)
How do we find the genes? (i)
(i) ORFs
Open reading frames, stretches of DNA that appear to code for protein sequence, begin with a Methionine (ATG).
Computer algorithms can predict the presence of a gene by searching for a transcription initiation site (TATA or GC box) sequence, ORFs, splice sites and other characteristics of a protein coding region
Most genes have multiple exons, and the final exon ends with a stop codon
Look for
-Met, TATA, three-codons, splice sites, end codons etc

How do we find the genes? (ii)
(ii) Transcriptome analysis
- Instead of sequencing genomic DNA, we can make a DNA copy of all the mRNA within a cell or tissue, and sequence that. If we collect all the different sequences we should end up with a representation of the transcriptome (genes that are being actively transcribed) of that particular cell or tissue.
- Based on the premise that if the cell is making mRNA from a gene then it must be useful to that cell, i.e. making a protein needed by that cell. The sequences are often called expressed sequence tags or ESTs.
mRNA to DNA with reverse transcriptase
How do we find the genes? (iii)
(iii) Comparative genomics
Line up sequences of related genomic DNA e.g. human vs. chimp and look for conserved (highly similar) regions
Regions that match up are important
Define bacteriophage
A type of virus that infects bacteria
What’s a contig?
Computer-assembled (virtual) region of genome sequence made by aligning overlapping smaller sequences reads
Genome - define
The complete sequence of an organism’s chromosomal DNA
Define genomics
The study of genomes, using methods to sequence (read) and assemble entire genomes and to analyse the data
Define ORF
Open Reading Frame - a region of DNA that looks like it would code for a protein or peptide
Define Transcriptome
Set of mRNAs transcribed by a particular cell or tissue, representing the genes that are coding for useful proteins in that cell or tissue
Okay, 7 key events in the history of genomics (time timeline) - what are they and when?
- 1977 - Fred Sanger manually sequences a 5.4Kbp bacteriophage
- 1995 - J. Craig Venter sequences first bacterial genome H. flu using “shotgun” sequencing. 1830Kbp
- 1998 - Human Genome Consortium (HGC) sequences first multicellular (eukaryote) organism, the nematode worm C. Elegans, 97Mbp
- 2001 - HGC completes Human genome sequence, 3.2 Gbp ($100M)
- 2007 - James Watson’s genome sequenced in two months with new technology - 6Gbp ($2M)
- 2012 - 1000 genome project completed ($7k)
- 2014 - Epi4K 4000 epilepsy genomes