Histopathology Flashcards
Blood supply of the liver
Dual blood supply: portal vein, hepatic artery
What are the cells of the liver?
Hepatocytes
Bile ducts
BVs
Endothelial cells
Kupffer cells
Stellate cells
What is the basic structure of the liver?
Hepatic lobule
At the centre are the terminal branches of the hepatic vein. The angles of the hexagon are formed by the portal tracts that contain 3 structures: BD, hepatic artery and portal vein
Where are centrilobular hepatocytes found?
Where are periportal hepatocytes found?
Located near the terminal hepatic vein i.e. zone 3: more metabolically active.
Located near the portal tract, receive blood rich in nutrients and O2.

What organ is this?

Liver
What are the functions of the liver?
Metabolic: involved in glycolysis, glycogen storage, glucose synthesis, amino acid synthesis, FA synthesis, lipoprotein metabolism. Drug metabolism.
Protein synthesis: make all circulating proteins except gamma globulins, including albumin, fibrinogen, and coag factors
Storage: glycogen, viamins A, D and B12 in large amounts. Small amounts of vitamin K, folate, Fe, Cu
Hormone metabolism: activates vit D. Conjugation and excretion of steroid hormones (oestrogens, GCs)< peptide hormone metabolism (insulin, GH< PTH)
Bile synthesis: 600-1000ml daily.
Immune function: antigens from gut reach liver via portal criculation and phagocytosed by kuppfer cells.
Which protein is not synthesised in the liver?
Gamma globulins
Which vitamins are stored in the liver?
A, D , B12 (large amount)
K (small amount)
What is this structure

Portal tract
What cellular changes occur following injury to the liver?
Loss of hepatocyte microvilli
Activation of stellate cells
Deposition of scar matrix
Loss of fenestrae
Kuppfer cell activation
Definition of cirrhosis
Involves whole liver
Fibrosis
Nodules of regenerating heaptocytes
Distortion of liver vascular architecture: intra and extra hepatic shunting of blood
How can cirrhosis be classified?
According to nodule size: micro or macronodular
According to aetiology: ETOH/insulin resistance, viral heaptitis etc
What are the complications of cirrhosis?
Portal HTN
Hepatic encephalopathy
HCC
What are the causes of acute hepatitis
Viruses
Drugs
What is the process shown here?
What is this known as?*

Acute hepatitis
“Spotty necrosis”
What are the causes of chronic hepatitis?
What does the grade of chronic hepatitis refer to?
Stage?
Viral
Drugs
Autoimmune
Grade= severity of inflammation
Stage= severity of fibrosis
What does this show?

Portal hepatitis
What does this show?
Also known as?

Interface hepatitis
“piecemeal necrosis”
What does this show?

Liver fibrosis
What is the pathological course of hep C infection
Acute: asymptomatic, 15-30% clear the infection
Fibrosis: (F0-F3), virus unlikely to clear without treatment, people may still be asymptomatic. Fatigue, URQ discomfort, transient appetite loss. End stage symptoms: itching, depression, impaired memory.
Scarring can be mild to severe. Extrahepatic Cxs: cryoglobulinaemia, glomerulonephritis, kerratoconjunctivitis sicca.
Cirrhosis (F4): Symptoms due to hepatic insufficiency and portal HTN: ascites, oesophageal varices, hepatic encephalopathy. Consider OLT
HCC: early stage: OLT, Sx, percutaenous ablation. Intermediate: TACE, Terminal
Spectrum of ETOHic liver disease?
Fatty liver
Alcoholic hepatitis
Cirrhosis
What does this show?

Fatty liver disease
What does this show?
What are the arrows pointing to?

Alcoholic hepatitis
Mallory bodies
What does this show?
What are the features?
Alcoholic liver cirrhosis
Micronodular
What does NAFLD look like?
What causes it?
Histologically looks like alcoholic liver disease
Due to insulin resistance associated with raised BMI and DM
What is PBC?
Features
Abs?
Primary biliary cirrhosis
Bile duct loss associated with chronic inflammation: granulomas
AMAs (anti-mitochondrial)
What does this show?

Primary biliary cirrhosis
Bile duct loss with granulomas
What is PSC?
What are the features?
With what is it associated?
Primary sclerosing cholangitis
Periductal bile duct fibrosis leading to loss
Associated with UC
Increased risk of cholangiocarcinoma
ERCP Dxic
What does this show?

Primary sclerosing cholangitis
What is haemochromatosis?
Cause?
Cxs?
Genetically determined increased gut iron absorption
Gene on chromosome 6
Parenchymal damage to organs secondary to Fe deposition-> “bronzed diabetes”
What does this show?

Haemochromatosis
What is haemosiderosis?
Accumulation of Fe in macrophages
Seen following blood transfusion
What is Wilson’s
By what is it caused
Cxs?
Accumlation of Cu due to feailure of excretion by hepatocytes
Genes on chromosome 13
Accumulates in the liver and CNS-> hepatolenticular degenration
What is a histopathological stain for Wilson’s?
Rhodanine
What disease is this?

Wilson’s wth a rhodanine stain.
What is a clinical sign in Wilson’s?
Keyser-Fleischer rings
What are the features of autoimmune hepatitis?
Interface hepatitis with plasma cells. Anti-SMA Abs
Responds to steroids.
What is Alpha-one antitrypsin deficiency?
Failure to secrete alpha-one antitrypsin
Leads to intra-cytoplasmic inclusions, hepatitis and cirrhosis

Alpha-1 antitrypsin
What are the causes of hepatic granulomas?
Specific: PBC, drugs
General: TB, sarcoid
What are the benign liver tumours?
Liver cell adenoma
bile duct adenoma
haemangioma
What is the most common type of liver tumour?
Secondary > primary
What are the primary liver malignancies?
HCC
Hepatoblastoma
Cholangiocarcinoma
Haemangiosarcoma
What does this show?

HCC
With what is cholangiocarcinoma associated?
Whence can it arise?
How is the prognosis?
PSC, helminth infection, cirrhosis, congenital liver abnormalities, Lynch syndrome Type II
Intrahepatic ducts, extrahepatic ducts (including GB)
Poor
What does this show?

Cholangiocarcinoma
What are the clinical features of hepatic adenoma?
Associated with OCP
Presents with abdo pain/ intraperitoneal bleeding
Resection if symptomatic, >5cm or if no shirnkage when stopping OCP
What are the features of haemangioma:
Most common benign lesion
No Rx
What are causes of HCC?
Ix?
Hepattis B + C, ETOHic cirrhosis. Haemocrhomatosis, NAFLD, aflatoxin, androgenic steroids.
Ix: alpha fetoprotein, USS
What is Lynch Syndrome caused by?
What are the classifications?
MMR defect
Type 1: HNPCC
Type 2: Extracolonic
What are the common sites to metastasise to the liver?
GI, breast or bronchus
What are the major causes of cirrhosis
ALD
NAFLD
Chronic viral hepatitis (B+/-D, C)
Autoimmune
Biliary causes:PBC and PSC
Genetic: haemochromatosis (HFE gene), Wilsons (ATP7B gene), A1AT, galactosaemia, glycogen storage disease
Drugs e.g. methotrexate
What are the causes of micronodular cirrhosis?
Uniform liver involvement (<3mm)
Alcoholic hepatitis, biliary tract disease
What are the causes of macronodular cirrhosis?
Variable nodule size (>3mm)
Viral hepatitis, Wilson’s, A1AT
What is the pathological process in cirrhosis?
Chronic inflammation causes stellate cell activation in the space of Disse
They become myofibroblasts that initiate fibrosis by deposition of collagen in space of Disse
Myofibroblasts contract, constricting sinusoids and increasing vascular resistance.
Undamaged hepatocytes regenerate in nodules between fibrous septa
What is the name of the score used to indicate Px in liver cirrhosis?
On what is it based?
Modified Child’s Pugh
Ascites
Encephalopathy
Bilirubin
Albumin
Prothrombin time
What are the thresholds for the Child pugh score?
<7: 45% 5 year survival
7-9: 20%
>10: <20%


What is portal HTN secondary to?
What happens?
Increased vascular resistance in liver
Hyperdynamic circulation
Sodium retention and plasma volume expansion
When portal pressure >10-12 mmHG, venous system dilates and collateral vessels form
Where are the collateral vessels found in portal HTN?
GORLDRA
GO junction
Rectum
L renal vein
Diaphragm
Retroperitoneum
Anterior abdominal:umbilical vein
What are the causes of portal HTN?
Pre-hepatic: Portal vein thrombosis: Factor V leiden
Hepatic
Pre-sinusoidal: Schistosomiasis, PBC, sarcoid
Sinusoidal: cirrhosis
Post-sinusoidal: veno-occlusive disease
Post-hepatic: Budd-Chiari syndrome
Causes of Budd-Chiari Syndrome
Mx of Budd-Chiari
30% idiopathic
Thrombophilia
OCP
Leukaemias
Compression by renal tumours, HCC
RTx
Thrombolyse, treat underlying cause. TIPS
What is a TIPS?
Transjugular Intrahepatic portosystemic shunt
What are the macroscopic and microscopic features of
Hepatic steatosis
Large, pale, yellow, greasy
Accumulation of fat droplets in hepatocytes
Fully reversible if ETOH avoided
What are the macroscopic and microscopic features of
Alcoholic hepstitis
Large, fibrotic liver
Hepatocyte ballooning and necrosis due to accumulation of fat, water and proteins
Mallory bodies
Fibrosis
Seen acutely after heavy night of drinking. Can range from asymptomatic to fulminant liver failure
What are the macroscopic and microscopic features of
Alcoholic cirrhosis
Yellow-tan, fatty, enlarged, transforms into shrunken, non-fatty brown organ
Micronodular cirrhosis (<3mm)
What are the features of NAFLD?
Hepatic steatosis in non-ETOHs
Most common cause of chronic liver disease in West
NAFLD: simple steatosis
NASH: steatosis and hepatitis, can progresss to cirrhosis
What are the features of autoimmune hepatitis?
Common with other autoimmune disease e.g. coeliac, SLE< RA, thyroiditis, Sjogrens, UC
78% female, young and postmenopausal
HLA-DR3
What is Type 1 autoimmune hepatitis?
ANA, anti-SMA< anti-actin, anti-soluble liver Ag
What is Type 2 autoimmune hepatitis?
Anti-LKM (liver, kidney, microsomal)
Mx of autoimmune hepatitis
Immunosuppression until transplant.
Disease recurs in 40%
Features of PBC
Epidemiology
LFTs
Abs
Hx
Mx
Auotimmune inflammatory destruction of medium sized intra-hepatic bile ducts-> cholesstasis->slow development of crrhosis
F>M 10:1
Peak incidence 40-50y/o
raised ALP, cholesterol, IgM, hyperbilirubinaemia (late)
AMA in >90%
US scan shows no bile duct dilatation
Histology: bile duct loss with granulomas
Fatigue, pruritus and abdo discomfort. Skin pigementation, anthelasma, steatorrhoea, Vit D malabsorption, inflammatory arthropathy
Ursodeoxycholic acid in early phase
Features of PSC
Epidemiology
Bloods
Dx
Cx
Inflammation and obliterative fibrosis of extrahepatic and intrahepatic bile ducts-> multi-focal stricture formation with dilation of preserved semgnets
M>F
Peak incidence at 40-50y/o
IBD associated (UC)
Raised ALP< several associated auto-Ig (p-ANCA)
US: bile duct dilatation
ERCP: shows beding of the bile ducts due to multifocal strictures
Increased incidence of cholangiocarcinoma
Haemochromatosis
Epidemiology
Pathophysiology
Histology
Signs/Symptoms
Ix
Treatment
Genetic
40-50y/o
Autosomal recessive: mutated HFE gene 6p21.3-> fe absorption which deposits in liver, heart, pancreas, adrenals, pituitarry, joints, skin-> fibrosis
Fe deposits in liver stains with Prussian blue
Skin bronzing (melanin deposition), DM, hepatomegaly, cardiomyopathy, hypogonadism, pseudogout
Ix: raised Fe, ferritin, transferrin sa >45%, decreased TIBC
Venesection
Desferrioxamine
30% with cirrhosis -> HCC
Wilson’s disease
Epidemiology
Pathophysiology
Histology
Signs/Symptoms
Ix
Treatment
v. rare
11-14y
Autosomal recessive: mutated ATP7B (Chr13) encodes Cu transporting ATPase-> decreased biliary Cu excretion and deposition in liver, CNS, iris.
Cu stains with Rhodanine stain, Mallory bodies and fibrosis on microscopy.
Liver disease: acute hepatitis, fulminant liver failure or cirrhosis.
Neuro disease: parkinsonism, psychosis, dementia
Decreased serum caeruloplasmin, decreased serum Cu, increased urinary Cu
Lifelong penicillinamine
A1AT
Epidemiology
Pathophysiology
Histology
Signs/Symptoms
Ix
Treatment
Autosomal dominant: A1AT accumulates in hepatocytes -> intracytoplasmic inclusions -> hepatitis. lack of A1AT in lungs -> emphysema
Intracytoplasmic inclusions of A1AT which stain with periodic acid Schiff
Kids: neonatal jaundice
Adults: emphysema and liver disease
Ix: redcued A1AT absent alpha globulin band on electrophoresis.
What are the functions of bone?
Mechanical: support and site of muscle attachment
Protective: vital organs and bone marrow
Metabolic: Ca reserve
What is the composition of bone?
Inorganic
Organic
Inorganic: (65%)
Ca hydroxyapatite (10Ca 6PO4 OH2)
body store for 99% of body Ca
85% of P, 65% Na and Mg
Organic: bone cells and protein matrix
What is the structure of bone medial to lateral?
Medulla, Cortex, Periosteum
What is the structure of bone proximal to distal?
Diaphysis
Metaphysis
Epihpyseal line
Epiphysis
Subchondral bone
Articular cartilage
What are the features of cortical bones?
Long bones
80% of bony skeleton
Appendicular
80-90% calcified
Mainly mechanical and protective
What are the features of cancellous bone?
Vertebrae and pelvis
20% of skelton
Axial
15-25% calcified
Mainly metabolic
Large surface
What type of bone is this?

Cortical
What type of bone is this?

Cancellous
What are the types/classifications of bone?
Woven/lamellour
Anatomically: flat/long bones
- intramembranous and anedochondral ossification
Trabecular (cancellous)/compact (cortical)
What are the arrows pointing at?


What is the function of osteoBlasts?
Build bone by laying down osteoid
What is the function of osteoclasts?
Multinucleate cells of macrophage family
Resorb bone
What are osteocytes?
Osteoblast like cells which sit in lacunae in bone
What is a lacuna?
In histology, a lacuna is a small space containing an osteocyte in bone or chondrocyte in cartilage.
The Lacunae are situated between the lamellae, and consist of a number of oblong spaces.


Draw a diagram showing the modulation of osteoclastogenesis
RANK is expressed on the surface of osteoclast lineage cells
RANKL expressed on MSCs of ostebolast lineage and on B and T Ls
When RANKL binds to RANK this causes osteoclast precursor cell to differentiate, increasing bone resorption
OPG competed with RANK for RANKL

How do tumour cells mediate local growth of tumour in bone?
Oncogene products produced by tumour cells metastasising to bone influence the bone cells to resorb bone and promote local growth of the tumour. This is mediated by the RANK /OPG signalling pathway.
What type of malignancy often causes more bone growth than destruction?
Prostate carcinoma
What is metabolic bone disease?
Disordered bone turnover due to imblanace of chemicals in body: vitamins, hormones, minerals
Net effect: reduced bone mass-> pathological #
What are the 3 main categroies of metabolic bone disease?
Non-endocrine e.g. age-related
Endocrine: e.g. Vit D, PTH
Disuse osteopenia
What are the histological characteristics in metabolic bone disease
Where must the biopsy be taken?
Static parameters: cortical thickness and porosity, trabecular bone volume, thickness, number & separation of tranbeculae
Biopsy from iliac crest
What is this showing?
What is the pink?

Trabecular (cancellous) bone
The trabecular bone, the grey is the marrow
What can be used to study the hisodynamic parameters of bone?
Fluorescent tetracycline labelling
What is the aetiology of osteoporosis?
1o: age, post-menopause
2o: drugs, systemic disease
What is the pathogenesis of osteoporosis?
High turnover?
Low turnover?
Pathogenesis: low intiial bone mass or accelerated bone loss can reduce bone mass below # threshold
90% of cases due to insufficient Ca intake and post-menopausal oestrogen deficiecny
High turnover: increased bone resorption
Low turnover: decreased bone formation
What are the influencing factors for osteoporosis?
Nutrition and social practices: ETOH, smoking, malabsorption, Vit C & DD
Endocrine
Immobilisation
Iatrogenic: corticosteroids, long term heparin or phenyotin therapy, casstration, XS thyroid therapy
What are the risk factors for osteoporosis?
Advanced age
Female
Smoking
ETOH
Early menopause
LT immobility
Low BMI
Poor Diet/malabsorption
Thyroid disease
Low testosterone
Chronic renal disease
Steroids
What is the UK societal impact of osteoporotic #s?
50% of patients cannot live independently post #
20% die
What is the presentation of osteoporosis?
Back pain and #
Classic #s:
wrist (Colle’s)
hip: NOF and intertrochanteric
pelvis
>60% vertebral #s are asymptomatic with compresion # usually in T11-L2
Ix in osteoporosis
Lab:
Serum Ca, P and ALP (usually N)
Urinary Ca
Collagen breakdown products
Imaging
Bone densitometry
What are the cut offs for bone densitometry?
T score: 1-2.5SD below normal peak bone mass= osteopenia
>2.5SD= osteoporosis
What is osteomalacia?
Defective bone mineralisation:
either Vit DD or PO4 deficiency
What are the sequelae of osteomalacia?
Bone pain/tenderness
#
Proximal weakness
Deformity
What does this show?
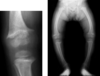
Osteomalacia (Rickets)
There is a widening of the growth plate which is also irregular and the femur and tibia become bowed as the child starts to walk and the legs have to weight bear.
What is the abnormality
What disease?

Horizontal # in Looser’s zone
Osteomalacia
Features of hyperPTHism
- Excess PTH
- increased Ca + PO4 excretion in urine
- hypercalcaemia
- hypophosphataemia
- skeletal changes of osteitis fibrosa cystica
What is the differnec between 1o and 2o hyperparathyroidism?
1o: parathyroid adenoma (85-90%) or chief cell hyperplasia
2o: chronic renal deficiency, Vit DD, malabsorption
Hypercalcaemic mnemonic
- Symptoms Mnemonic
- Stones (Ca oxalate renal stones)
- Bones (osteitis fibrosa cystica, bone resorption)
- Abdominal groans (acute pancreatitis)
- Psychic moans (psychosis & depression)
What is renal osteodystrophy?
Comprises all the skeletal changes of chronic renal disease
Increased bone resorption: osteitis fibrosa cystica
Osteomalacia
Osteosclerosis
Growth retardation
Osteoporosis
What does this XR show?

Osteitis fibrosa cystica: increase bone resorption
Lab features of renal osteodystrophy
- PO4 retention – hyperphosphataemia
- Hypocalcaemia as a result of decreased vit D
- 2o hyperparathyroidism
- Metabolic acidosis
- Aluminium deposition
What is Paget’s?
What are the 3 phases?
Disorder of bone turnover
Osteolytic
Osteolytic-osteosclerotic
Quiescent osteosclerotic
What is this?

Paget’s disease of bone
Features of Paget’s?
Onset >40y
M=F
Rare in asians/africans
Mono-ostotic in 15%, remained polyostotic
Aetiology is unknown
Familial pattern shows autosomal pattern with incomplete penetrance
Parvomyxovirus type particles have been seen in Pagetic bone
What are the 5 most commonly affected sites in Paget’s?
Vertebrae
Skull
Pelvis
Femur
Tibia
Clinical features of Paget’s?
Pain
micro#
Nerve compression
Skull changes may put medulla at risk
+/- haemodynamic chanes, HF
Development of sarcoma in area of involvement in 1%
What does this show?

Paget’s disease affecting tibia
What are the indications for bone biopsy?
Suspected osteomalacia
Diagnostic classification of renal osteodystrophy
Osteopaenia: in young patients (<50) or associated with abnormal Ca metabolism
Classification of hereditary childhood bone disease
XR. histological and biochemical findings for Osteoporosis
No XR
Loss of cancellous bone
N Ca, N PO4, N ALP
XR and histological findings for Osteomalacia
Looser’s zones: pseudo#s, splaying of metaphysis
Excess of unmineralised bone: osteoid
N/L Ca, L PO4, H ALP
XR and histological findings for 1o hyperparathyroidism
Brown’s tumours, Salt and pepper skull, Subperiosteoal bone resorption in phalanges
Osteitis fibrosa cystica: marrow fibrosis and cysts aka Brown tumour
Raised Ca,, Low/N PO4, Raised/N ALP
What are Brown’s tumours?
The brown tumor is a bone lesion that arises in settings of excess osteoclast activity, such as hyperparathyroidism. It is not a true neoplasm, as the term “tumor” suggests; however, it may mimic a true neoplasm.[1]Brown tumours are radiolucenton x-ray.

Hyperparathyroidism: Brown’s tuour of the hands

Salt and pepper sign of the calvaria refers to multiple tiny hyperlucent areas in the skull vault caused by resorption of trabecular bone in hyperparathyroidism.
There is loss of definition between the inner and outer tables of the skull and a ground-glass appearance as well as spotty deossification.
XR and histological findings for 1o Paget’s?
Mixed lytic and sclertoic. Skull: osteoprorosis circumscripta, cotton wool.
Vertebrae: picture frame, ivory vertebrae
Pelvis: sclerosis and lucency
Huge osteclosts w >100 nuclei, mosaic (like jigsaw) pattern of lamellar bone
N Ca + PO4, +++ALP

Osteoporosis circumscripta cranii (also known as osteolysis circumscripta) refers to discrete radiolucent regions of the skull on plain radiographs. They are often seen in context of the lytic (incipient-active) phase of Paget’s disease of the skull, but may be observed in other circumstances as well, e.g. hyperparathyroidism, leontiasis ossea 9.

Cotton wool skull- Pagets

Picture frame vertebra
Paget’s

Ivory vertebra
Paget’s
What are the features of astrocytes?
Most abundant
Anchor neurones by numerous projections, regulate environment e.g. ions, neurotransmitters and form BBB
Features of oligodendrocytes?
Coat axons with their cell membranes forming myelin
Features of ependymal cells?
Line the cavities of the CNS, make up walls of the ventricles, create and secrete CSF and beat cilia to help move the CSF.
Act as neuronal stem cells
Radial glia features
Arise from the neuroepithelial cells in embrogenes, act as the scaffold for new neurones
Features of Schwann cells?
Provide myelination to PNS
Features of satellite cells?
Small cells that surround the neurones in sensory sympathetic and parasympathetic ganglia, helping to regulate the environment
Features of enteric glial cells
Intrinsic ganglia of the GIT
Function of microglia
Act as macrophages
What are the glial cells?
Astrocytes
Oligodendrocytes
Microglia
Which cells interface with the CSF?
Ependyma
Choroid plexus epithelium
Meninges
Which CNS cells interface with blood?
Endothelium and pericytes
What is the most common CNS tumour?
Metastatic neoplasm
What are the principle malignancies causing neurometastases?
Leukaemias and lymphomas, more so in young.
Lung, breast and malignant menaloma
What are the features of brain mest?
Pathology?
Symptoms?
May involve the meninges as well as the parenchyma
Well demarcated solitary or multiple lesions with surrounding oedema
Neurological efffects and raised ICP
What are the 5 most common malignancies leading to CNS tumours in adults?
BLCPB
Breast
Lung
Large Bowel
Prostate
Bladder
What are the most common group of CNS primary tumours?
Astrocytomas
Astrocytomas
Age of onset
Pathology
Grading
Symptoms
Px
Any age, elderly usually worse
An infiltrative growth pattern in the cerebral hemispheres
Grading is based on the degree of dy/dx with histological grade an important predictor of behaviour
Raised ICP and focal neuro signs
Depends on site, grade and age of the patient
What are the types of astrocytoma?
Pilocytic astrocytomas: more common in children but can occur any age.
Anaplastic astrocytoma
Glioblastoma multiforme: necrotic, poorly differentiated tumour
Features of oligodendroglioma
Px
Most common in adulthood
Usually in the cerebral hemisphere and are soft and gelatinous
Better demarcated than infiltrating astroctomas
Calcifcaition common
Px less predictable, dependant on grade site, patient age, cytogenetics etc
Features of ependyomas?
Symptoms?
NB AA?
Occur at any age
Most offen within the ventricular cavities or within the canal of the SC
Usually well demarcated
Symptoms depend on site: intracranial: hydrocephalus or raised ICP
Anaplastic astrocytomas/variants often also disseminate through the sub arachnoid space
Where are ependyomas found in <20y/o?
In adults?
In te ventricular cavities
In the SC
Features of primitive neuroepithelial neoplasms
Composed of embryonal primitive cells
Most common in children
Undifferentiated lesions
Px: most survive 5y or more
Medulloblastoma features
Lesion of the cerebellum in the first 2 decades of life
Leads to raised ICP and cerebellar signs
Features of primary CNS lymphoma
Increased since AIDs
Features of meningiomas
Derive from meningioethlial cells
Most occur in the brain parenchyma but can occur in the cranial vault and cord.
Usually adults
Increased in NF2
Usually lobulated lesions, sharp interface between tumour and parenchyma
Overlying skull may be thickened or invaded by the tumour
Symptoms: raised ICP with focal neurological signs
NF2 in brain tumours?
Meningioma
Ventricular tumour/hydrocephlaus in brain tumours?
Ependyoma
Indolent/childhood in brain tumours?
Pilocytic astrocytoma
Soft gelatinous calcified brain tumour
Oligodendroma
What genetic conditions predispose to CNS malignancy?
VHL
NF 1 + 2
Li-Fraumeni
Gorlin
Turcot syndrome
Sings of supratentorial tumours
Focal neurological deficit
Seizure
Altered mental status
Headache
Signs of subtenotrial tumours
Cerebellar ataxia
Long tract signs
CN palsy
What are the neurosurgical approaches to a CNS malignancy
Stereotacic biopsy
Open biopsy
Craniotomy for debulking
CNS tumours:
Grade 1
2
3
4
LT survival
Causes death in >5y
Death in <5y
Deaith within 6m-1y
Features of infiltrative gliomas
Account for 80% of gliomas.
Astrocytomas- Oligodendrogliomas and mixed
What is the most aggressive infiltrative glioma?
De novo glioblastoma
What mutation is responsible for 80% of diffuse astrocytomas?
IDH1
What is secondary glioblastoma?
From progression of a lower grade astrocytoma
What is the proportion of diffuse astrocytomas found in the crebellum?
10%
Which has the better Px, astrocytomas or oligodendroglioma?
What is important?
Oligodendroglioma
Resection
- Usually 1st and 2nd decade - 20% of CNS tumours below 14 years and 15% between 14-18 years
- Often cerebellar, optic-hypothalamic, brain stem
- Often cystic. Always contrast enhancement
- They can disseminate in the subarachnoid space (es: follow nerve roots)
- Compressive margins (never diffuse infiltration)
- Variable histological features
- Very often Rosenthal fibres and granular bodies
- Hallmark: Piloid “hairy” cell
Pilocytic astrocytoma
- Rare (0.5 per 100,000 year, in children)
- 75% arise in the vermis in children and hemispheric in adults
- Present with cerebellar signs, cranial hypertension
Medulloblastoma
- 24-30% primary intracranial tumours
- Incidental in up to 10% of post-mortem
- Usually adults – rare in patients younger than 40 (more aggressive)
- Focal symptoms (seizure, compression)
- Any site of craniospinal axis
Meningioma
What is crucial in the assesmnet of grade of CNS primaries?
Mitotic activity
What is occuring in this image?

Pseudoinvasion along Virchow-Robin’s space
What is the commonest cause of CNS disease?
What is the most common cause of this?
Infarction
Cerebral atherosclerosis
What proportion of strokes are infarctive?
70-80%
Where do thrombosis occur in the CNS?
Atherosclerosis affects the larger extracerebral vessels worse, often near the carotid bifurcation or in the basilar artery
Which part of the CNS do emboli affect?
Intracerebral atery, usually from the heart or atherosclerotic plaques
Usually occurs in the MCA branches
What is a watershed infarction?
Occurs on border zone, hypoperfusion of the most distal edges of the blood supply. Not necessarily occlusive.
ACA and MCA most at risk
Site: distally is affected more than proximal
Dependant on the anastamoses around the zone
Cortical border zone
Between ACA and MCA
Internal border zone?
Between LCA and MCA
Cortical border zone
Between MCA and PCA
Def: TIA
Under 25hrs, self-limiting vascular obstruction, emboli and or platelet-fibrin aggregates
Px of TIA
1/3rd will get a significant infarct within 5 years
Def: intraparenchymal haemorrhage
Presentation
Site
Haemorrhage into the brain substance, usually rupture of one of the small intraparenchymal vessels
Raised ICP, rapid LOC
Basal ganglia, abrupt onset
Cause of ICH
50% HTN
Weakening of the walls, accelerated atheroscleroris in LV, hyaline atherosclerosis in smaller vesseles.
Clotting disorders, neoplasms, amyloid, vasculitis and vascular malformations also contribute
SAH most common cause
Most cmmon sites
Rupture of Berry aneurysm (present in 1% of the population)
80% ICA bifurcation, 20% vertebrobasilar circulation
30% of patients they are multiple
Berry aneurysm
>6-10mm
>25mm
Greatest risk of rupture
Mass lesion
Presentation and Px of SAH
Sudden onset, severe headache, vomiting, LOC
50% die within a few days, worse Px if there were warning bleeds
Types of vascular malformations
Significance
AVM
Capillary telangectasias
Venous Angiomas
Cavernous angiomas
Important cause of ICH and developmental abnormalities
EDH features
Meningeal artery rupture which lies between the dura mater. Associated with skull fractures
Presentation of EDH
Lucid interval followed by progressive LOC. Bleeding is arterial and there is subsequent mass effect
SDH features
Between the dura and the arachnoid mater.
Disruption of the bridging veins.
Associated with rapid change of head velocity which leads to tearing of the veins.
Usually clear Hx of tumour caused by raised ICP. Venous bleeding therefore slower development than EDH.
Presentation of SDH
There is a less good history of trauma and it is associated with brain atrophy. Often causes vague alteration in mental state rather than classical features of raised ICP
Features of Parenchymal injury
Occurs due to sudden acceleration or deceleration with sufficient force to tear nerve cell process in the cerebral white matter
Presentation of parenchymal injury
Sequelae?
Concussion, transient LOC and neurological deficit. Sometimes with seizrues with recovery over hours or days.
Diffuse axonal injury: causes most post-traumatic dementia and with hypoxic ischaemic injury is the leading cause of persistent vegetative state
Coup vs contracoup
Contustion at site vs away from site of impact
Need to know the MOI
What is Duret’s Haemorrhage
Bleeding in the ventral and paramedian part of the upper brainstaim (pons and midbrain)
Features of cerebral oedema
Gyral flat and te sulci are obliterated
Causes of brain oedema
Anything that can damage the BBB
Can be classified as
Vasiogenic: where the integrity of the BBB is disrupted which is aggravated by the lack of lymphatic CNS drainage
Cytotoxic: secondary to cellular injury e.g. due to general hypoxic ischaemic injury.
Features of herniation
Due to raised ICp, can force the brain against unyielding bony wall
Subfalcine herniation
Cingulated gyrus displaced under falx cerebri
Transtentorial hernia
Uncal gyral, medial temporal lobe compressed against the free margin of the tentorium cerebelli
Results in compression of the PCA and oculomotor nerve
Tonsilar herniation
Cerebellar tonsils herniate through the foramen magnum causing brainstem compression
Stroke
Symptoms
Vascular territories
Ix
Mx
Sudden onset, FAST, numbness, loss of vision, dysphagia
MCA most common
CT/MRI (infarct vs haemorrhage)
Ix for vascular risk: BP, FBC, ESR, U&E, glucose, lipids, CXR, ECG, carotid doppler
Aspirin +/- dipyridamole
Thrombolytics (<3h)
+/’- caroit dendarterectomy
LT: treat HTN, reduce lipids, anticoagulate
TIA
Symptoms
Vascular territories
Ix
Mx
<24hrs, amaurosis fugax, carotid bruit
Any however characteristically embolic atherogenic debris from the carotid artery travels to the opthalmic branch of the internal caroitd
Cartoid USS
IX for vascular risk as in stroke
As for stroke except no thrombolysis
What proportion of SAH are from ruptured Berry Aneurysm
What conditions increase risk of Berry Aneurysm
85%
PKD, Ehler’s Danlos and patients with coarctation
Also associated with AVMs, capillary telangectasias, venous and cavrnous angiomas
Haemorrhage brain injury classification
Non traumatic:
ICH
SAH
Traumatic:
EDH
SDH
6 types of brain herniation
Uncal
Central (transtentorial)
Cingulate (subfalcine)
Transcalvarial
Upward
Tonsilar

Cerebral oedema
What are the two types of hydrocephalus?
Communicating/non-obstructive
Non-communicating/obstructive
Causes of non-obstructive hydrocephalus
Impaired reabsorption of CSF:
Normal pressure hydrocephalus
Hydrocephalus ex-vacuo
Features of normal pressure hydrocephalus
Associated with elevated CSF causing increased ICP and increased ventricular size
Thought to be scondary to an increase in fluid levels.
Infection, tumours, trauma and haemorrhage
Features of hydrocephalus ex-vacuo
Atrophy causing secondary enlargement of the ventricle and subarachnoid space
Not the result of increased CSF production pressure but a compensatory enlargement of ventricle due to loss of brain parenchyma
Causes of obstructive hydrocephalus
Actual obstruction to CSF flow
Adhesion, external compression, interventricular cysts and tumours
Def of stroke
This excludes
A stroke is a clinical syndrome characterised by rapidly developing clinical symptoms and / or signs of focal, and at times global loss of cerebral function, with symptoms lasting more than 24 hours or leading to death, with no apparent cause other than that of vascular origin (Hatano, 1976).
This definition includes stroke due to cerebral infarction, primary intracerebral haemorrhage, intraventricular haemorrhage, and most cases of subarachnoid haemorrhage
It excludes subdural haemorrhage, epidural haemorrhage, or intracerebral haemorrhage (ICH) or infarction caused by infection or tumour.
Two types of cerebral ischaemia
Focal: defined bascular territory
Global: failure of systemic circulation
NB re atypical brain infarct
Atypical refers to odd anatomy – not classical vascular territory
Clinical features and neuroimaging can be misleading.
“Atypical brain infarct may mimic tumors”
Mx of AVM
Surgery, embolisation, Radiosurgery
Features of AVM
- Occur anywhere in the CNS
- Becomes symptomatic between 2nd and 5th decade (mean age 31.2 years)
- Present with haemorrhage, seizures, headache, focal neurological deficits
- High pressure – MASSIVE BLEEDING!!!
- Seen at angiography
- Risk of bleeding 1.3-3.9% yearly
- Risk of re-bleeding 6.0-6.9% during the first year
- Morbidity after rupture 53-81% - high in eloquent areas
- Mortality 10-17.6%
Features of cavernous angioma
“Well-defined malformative lesion composed of closely packed vessels with no parenchyma interposed between vascular spaces”
Anywhere in the CNS
Usually symptomatic after age 50
Pathogenesis unknown
Rarely multiple – familial (linkage chrom. 7p, 7q, 3q)
Present with headache, seizures, focal deficits, haemorrhage
A few feeding vessels
Low pressure – RECURRENT BLEEDINGS
Angiographically usually negative
Therapy: surgery
Histopathological differentiation between infarct and haemoorhage
INFARCT
- Tissue necrosis (stains)
- Rarely haemorrhagic
- Permanent damage in the affected area (except for penumbra)
- No recovery
HAEMORRHAGE
- Bleeding
- Dissection of parenchyma
- Less macrophages
- Limited tissue damage (periphery)
- Partial recovery
Lesions in fatal non-missile head injury
Primary
Skull fracture 75%
Surface contusions 95%
Diffuse axonal injury (DAI) 30%
Intracranial haematoma 60%
Secondary
Brain swelling 53%
Ischaemic brain damage 55%
Infection 4%
Due to raised ICP 75%
Intracranial haematoma
66% of fatal non-missile head injury
10% extradural
56% subdural, subarachnoid, intracerebral or burst lobe
surgical evacuation



SDH

EDH


What are connectomes?
Functional conective areas in the brain
What are the central neurotransmitters
GABA: inhibitory
Glutamate: excitatory
Ach, dopamine, serotonin
What are microglial cells?
Resident macrophage population constituting 20% CNS cells. Have motile processes. Involved in immune surveillance and tissue reomdelling (synaptic stripping)
Cytotoxic in neurodegeneration and neuroinflammation
Def: neurodegenerative disease
Progressive, irreversible condition leading to neuronal loss
Often caused by intra or extracellular accumulation of a misfolded protein. Usually sporadic resulting in dementia.
Common in the ageing population
Def: neuroinflammatory disease
Conditions characterised by an innate and/or adaptive inflammatory response. Can be reveersible, progressive irreversible
Def: dementia
A serious loss of global cognitive ability in a previously unimpaired person beyond what might be expected from normal ageing. Can be a static event e.g. from TBI or due to neurodegeneration.
Dx of dementia
Difficult to diagnose on symptoms alone, some have a distinct clinical phenotype which gives a high probability of a dx e.g. PD.
Based on neuroimaging, brain biopsy or PM examniation
Exmaples of misfolded proteins implicated in dementias?
Tau, beta-amyloid, alpha-synuclein, Huntingtin, PrP, Fus
Features of AD
Dense deposits around neurones (neuritic plaques)
Twisted bands of fibre (NFTs)
Begins after 5th-6th decade, pathogenesis unclear.
Pathological findings in AD
Senile plaques: complex spherical structures involving the grey matter.
Diffuse plaques are amorphous and seen in ageing brains.
Neuritic plaques consist of clusters of radially orientated abnormal axons and dendrites= dystrophic dendrites
Features of NFTs
Ubiquitin abnormally accumulates inside cells, indicates a disease process. These protein accumulations= inclusion bodies
NFTs are aggregates of hyperphosphorylated Tau proteins.Intracellular structures composed of two filaments wound in a double helix
What conditions features Tau?
AD, progressive SNP, some other uncommon conditions
Features of Tau
Normal component of the neuronal cytoskeleton, stabilises MTs. 6 isoforms.
Function is regulated by kinases.
Unphosphorylated or hyperphosphorylated Tau fails to bind to MTs, accumulating in NFTs and neuritic plaques

Senile plaque

NFT
Risk factors for AD
Age
FHx
T21
Head trauma
PD and depression
Dx of AD
Clinical suspicion
MRI and PET scans helpful
Role of biopsy unclear
Dx and PM is definitive
Features of Beta amyloid
Derived from APP which is a ubiquitously expressed membrane molecule.
Cleaved by secretases which are regulated by Presenilin 1
Amyloid accumulation results from defective APP cleavage

Beta amyloid
What fragment of amyloid accumulates in AD?
Fragment 39-42
What is the role of ApoE in AD?
Produced by astrocytes and mediates phospholipid and cholesterol mobilisation
ApoE4 has a high affinity to beta amyloid and less activity.
ApoE4 is a risk factor for late onset sporadic AD
Rx in AD
Classes, e.g.
Acetylcholinesterases: Tacrine (Cognex(), Donepezil, Rivastigmine. Produce mild benefits without influencing progression
nAChRs: galantamine
Glutamate antagonist: memantine (also used in vascular dementia)
DDx in Parkinson
Parkinsonian syndromes:
Primary: PD
Secondary
Parkinson-plus syndrome
Familial Neurodegenerative diseases
Primary parkinsonism=
PD (sporadic, familial)
Causes of secondary parkinsonism
Drug induced: dopamine antags and depletor
Hemiatryophy-hemiparkinsonism
Normal pressure hydrocephalus
Hypoxia
Infectious: postencephalitic
Toxin: e.g. MPTP
Trauma
Tumour
Vascular: multiinfarct state
Causes of parkinson plus syndromes
Cortical-basal ganglionic degeneration
Dementia sydnromes: AD, LBD, frontotemporal dementia
Lytico-Bodig (ALS)
Multiple system atrophy syndromes: Shy Drager, OPCA, MND parkinsonism
Progressive pallidal atrophy
PSN Palsy
Familial Neurodegenerative diseases causing parkinsonism
Hallervoden-Spatz
Huntingoton
Lubag
Mitochondrial cytopathies with Striatal Necrosis
Neuroacanthocytosis.
WIlsons
Features of PD
Usually sporadic, develops after 50y
Pathogenesis unclear
Symptoms of PD
Attributed to cell death of the dopamine producing neurones in the SN
Need to lose 80-85% of the dopaminergic neurones and deplete dopamine levels by 70% before they appear.
Cell death in substantita nigra results in depigmentation
Lewy Bodies found in cell bodies and Lewy neurites in neuronal projections
Alpha-synuclein and ubiquitin positive
Clinical features of PD
Resting tremor, rigidity, bradykinesia, autonomic dysfunction, dysphagia
Psychiatric: hallucinations, anxiety and dementia
L-DOPA responsiveness
What is alpha-synuclein
Small protin associated with synaptic membrane, accumulates and has toxic effect.
Mutations reported in instances of familial parkinson’s

Lewy Bodies
Causes of demyelinating lesions
Viral infections: PML (progressive multifocal leukoencephalopathy caused by JC virus in immune deficiencies
Genetic: leukodystrophies
Autoimmune: MS, acute haemorrhagic encephalomyeltisi, acute disseminated encephalomyelitis
Nutritional: central pontine myleniosis
Function of myelin
Produced by oligodendrocytes, fundamental for axon conduction, highly susceptible to damage.
Lipid rich insulating membrane
Features of MS
Peak age: 20-40y
Assocaited with some HLAs
Usually presents with focal symptoms, optic neuritis and poor coordination
What are the types of MS
Relapsing remitting: recovery less severe, evolves into secondary progressive agter years
Primary progressive (10%): no recovery after episodes of demyelination: severe
Progressive relapsing
What is neuromyeltitis optica
Rare variant of MS
Aka Devic disease
Autoimmune disease against the SC and optic nerves with auqaporin 4 attacked
What is Balo disease?
Rare variant of MS
Rapidly progressive with concentric scoliosis
Pathology of MS
Loss of myelin, lesions centred by veins with sharpy margins
Axonal preservation in early plaques
Active plaques- glial scar
Remyleinating shadow plaques
Infiltration of macrohpages.
Cortical involvement is the leading cause of disability, CI leading to dementia, related to inflamm rather than demyelination
Patholocial protein in:
AD
Tau, beta-amyloid
Patholocial protein in:
LBD
Alpha synuclein, ubiquitin
Patholocial protein in:
Corticobasal degeneration
Tau
Patholocial protein in:
Frontotemporal dementia
Tau
Patholocial protein in:
Pick’s disease
Tau
Imaging findings in AD
Generalised atrophy of the brain
Widened sulci
Narrowed Gyri
Enlarged ventricles (most marked in the temporal and frontal lobes with loss of cholinergic neurones)
Features of LBD
Psychological disturbances occur early, day to fluctuations in cogntiive performance
Vsiual hallucinations
Spontaneous motor signs of Parkinsonism
Recurrent falls and syncope
Myelin basic protein and proteo-lipid protein=
MS
Features of multiple system atrophy
Types
Degenerative neurological disorder characterised by features that can present in a similar manner to Parkinson’s but show a poort response to parkinson’s medication
Shy Drager
Stratonigral
Olivopontocerebellar
Shy Drager
MSA: autonomic dysfunction
Striatonigral MSA
Difficulty with movement
Olivopontocerebellar MSA
Difficulty with balance and coordination
Features of vCJD
vSporadic neuropsychiatric disorder
vPatients <45 yrs old
vCerebellar ataxia
vDementia
vLonger duration than CJD
vLinked to BSE
Neuropathology of AD
- Extracellular plaques
- Neurofibrillary tangles
- Cerebral amyloid angiopathy (CAA)
- Neuronal loss (cerebral atrophy)

Cortical atrophy- AD
Diagnotics gold standard in PD
alpha-synuclein immunostaining
Functions of the kidney
Excretion of metabolic waste products and foreing chemicals
Regulation of fluid and electrolyte balance, acid base balance
Hormone secretion: EPO, Renin and 1,25 cholecaliferol
What are the two types of glomerulaur disease
Failure to:
Filter an adequate amount of blood resulting in a lack of waste product excretion
Failure to maintain a barrier function leading to the loss of protein and/or blood cells in the urine
What must be distinguished in renal disease
Syndrome: e.g. ARF, nephrotic etc.
Morphological changes: glomerulonephritis, thrombotic microangiopathy
Aetiologies: SLE, amyloidosis, drugs and infetions
What are immune complexes in the context of renal disease?
Antigens
Rate
Stie
Composed of a lattice work of Ab and Ag, may become deposited in te glomerulus and lead to an inflammatory response-> complement activation and stimulation of inflammatory cells through Fc receptors
May be endogenous e.g. SLE or exogenous e.g. derived from an infective organism
Rate: occur at different rates: rapidly progressive glomerulonephritis vs slow onset
Site may bary but glomerular injury is determined by the immune complex location
Congenital diseases of the kidney
Bilateral/unilateral agenesis
Ectopic e.g. pelvic
Horseshoe (usually fused at the lower pole)
Features of Adult PKD
Causes 10% of ESRF
Cysts arise from all portions of the nephron
Renal failure develops from 40-70y
Genes: PKD1 and PKD2
Features of acquired cystic disease
Cysts develop in the kidneys of ESRF on dialysis
Carcinoma can develop in 7% in 10y
What is acute renal failure?
What does it result in?
Biochemically?
Rapid loss in golmerular filtration and tubular function
Results in an abnormal water and electrolyte balance
Reduced GFR manifestated as rasied serum creatinine (Normal= <1.3) and urea (10-20mg/dL)
May r3esult in acidosis, hyperkalaemia and fluid overload
How can disease of the kidney be classified?
e.g.?
According to the site of the nephron it affects
- Glomerulus
Nephrotic syndrome
Nephritic syndrome
- Tubules and interstitium
ATN
Tubulointerstitial nephritis: acute phyelonephritis, chronic pyelonephritis & refulx nephropathy, interstitial nephritis
- BVs:
Thombotic microangiopathies (HUS, TTP)
Features of nephrotic sydnrome
Proteinuria (>3g/24h)
Hypoalbuminaemia
Oedema
(+hyperlipidaemia)
“Swelling” facial in children, peripheral in adults
“frothy urine”
What are three primary causes of nephrotic syndrome
Minimal change disease
Membranous glomerular disease
Gocal segmental glomerluosclerosis
Features of minimal change disease
Light microscopy
Electron microscopy
Immunofluorescnece
Px
Response to steroids
Primary cause of nephrotic syndrome
Most common in children (75%) with second peak in elderly
No changes
Loss of podocyte foot processes
No immune deposits
<5% ESRF
90% respond
Features of membranous glomerular disease
Light microscopy
Electron microscopy
Immunofluorescnece
Px
Response to steroids
Primary cause of nephrotic syndrome
Common in adults (30%)
Diffuse glomerular BM thickening
Loss of podocyte foot processes. Subepithelial deposits spikey
Ig and complements in granular deposits along entire GBM
40% ESRF at 2-20y
Can be 1o or 2o to SLE, infections, drugs and malignancy
Poor response to steroids
Features of Focal Segmental Glomerulosclerosis
Light microscopy
Electron microscopy
Immunofluorescnece
Px
Response to steroids
Common in adults (30%), Afrocarribean
Focal and segmental glomerular consolidation and scarring. Hyalinosis
Loss of podocyte foot processes
Ig and complement in scarred areas
50% ESRF in 10y
1o but can 2o to obesity and HIV nephropathy
50% respond
What are 2 secondary causes of nephrotic syndrome
DM
Amyloidosis
Histology of DM nephrotic syndrome
Hints in question
Diffuse GBM thickening
Messangial matric nodules aka Kimmelstiel Wilson nodules
Asian
Histology of amyloidosis nephrotic syndreom
Hints
Apple green birefringence with congo Red
May have chronic inflammation:
RA, chronic infection.
Causes AA protien depositions
May have Ig light chain deposition most commonly from MM
Clinical clues of amyloidosis: Macroglossia, HF, hepatomegaly
Features of nephritic syndrome
Manifestation of glomerular inflammation i.e. glomerulonephritis
Haematuria
Dysmorphic RBCs and RBC casts in urine
May also have:
Oliguria
Raised urea and creatinine
HTN
Proteinuria (not in nephrotic range)
Causes of nephritic syndrome
Acute postinfectious
IgA Nephropathy (Berger Disease)
Rapidly progressive (crescenteric) GN
Hereditary nephritis (Alport’s)
Thin BM Disease (Bening familal haematuria)
What are the features of Acute Postinfectious GN
Occurs 1-3w post streptococcal throat infection or impetigo (usually Group A S= Strep pyogenes)
Glomerular damage due to immune complex deposition
Haematuria (red cell casts), proteinuria, oedema, HTN
Bloods: raised ASOT titre, decreased C3
Light microscopy: Increased cellularity of glomeruli
FM: granular deposits of Ig Ga and C3 in GMB
EM: subendothelial humps
Findings in Acute Postinfectous GN
Features of Berger disease
IgA Nephropathy
Most common GN worldwide
IgA complex deposition in glomeruli
Presents 1-2d after URTI with Frank haematuria
Main symptoms are persistent or recurrent frank haematuria or asymptomatic macroscopic haematuria
Can rapidly prgoress to ESRF
Biopsy: granular deposition of IgA and complement in mesangium
GN 1-2d after URTI with haematuria
IgA Nephropathy (Berger)
Nephropathy 1-3w post streptotoccal infection
Acute Postinfectious GN
Feature of Rapidly progresswive (Crescenteric GN)
Most aggressive GN causing ESRF within weeks
Presents as nephritic syndrome but oliguria and renal failure are more pronounced
How is cresenteric GN calssified?
Based on immunological findings:
Type 1: Anti-GBM Ab
Type 2: Immune complex
Type-3: pauci-immune/ANCA associated
Regardless of cause all are characterised by crescenets in glomeruli on light micrscopy
Features of T1 crescenteric GN
Cause
Light micrscopy
Fluorescnece microscopy
Additional organ involvement
Anti-GBM Ab
Goodpasture’s. HLA-DRB1 association
Crescents
Linear deposition of IgG in GBM
Lungs: pulmonary haemorrhage
Features of T2 crescenteric GN
Cause
Light micrscopy
Fluorescnece microscopy
Additional organ involvement
Immune complex mediated
SLE, IgA nephropathy, postinfectious GN
Crescents
Granular IgG immune complex deposition on GBM/mesangium
Often limited, except in SLE
Features of T3 crescenteric GN
Cause
Light micrscopy
Fluorescnece microscopy
Additional organ involvement
Pauci-immune i.e. lacking anti-GBM or immune complex
c-ANCA: Wenger’s granulomatosis
p-ANCA: microscopic polyangitis
Cresecents
Lack/scanty immune complex deposition
Vasculitis: skin rashes or pulmonary haemorrhage
Features of Alport’s
Hereditary Nephritis
Caused by mutation in Type IV collagen alpha 5 chain
X-linked
Nephritic syndrome + sensorineural deafness and eye disorders
Presents at 5-20y with nephritic syndrome progressing to ESRF
Features of Benign Familial Haematuria
Thin BM disease nephirits
Rarely causes nephritic syndrome, normally exclusively asymptomatic haematuria
Diffuse thinning of the GBM caused by autosomal dominant mutation in type 4 collagen alpha 4 chain.
Usually asymptomatic and diagnosed incidentally
Usually normal renal function
Differentials for asymptoamtic haematuria
Benign Familal Haematuria
Berger
Alport
NB: IgA and Thin BM are more common causes of asymptomatic haematuria than of nephritic syndrome. IgA more likely to cause frank haematuria and change in renal funciton and slighlty more common in Asian pop
Features of ATN
Histologically
Damage to tubular epithelial cells-> blockage of tubules by casts-> reduced flow and haemodynamic changes-> acute renal failure
Necrosis of short segments of tubules
What is the most common cause of acute renal failure?
ATN
What are the common causes of ATN?
Ischaemia: burns, septicaemia
Nephrotoxins: drugs (gentamicin, NSAIDs, radiographic contrast agents, mygolobin, heavy metals)
What is tubulointerstitial nephritis?
Causes?
A group of renal inflammatory disorders involving the tubules and interstitium
Acute pyelonephritis
Chronic pyelonephritis and reflux nephropathy
Acute interstitial nephritis
Chronic interstitial nephritis/analgesic nephropathy
What is acute pyelonephritis?
Bacterial infection of the kidney, usually as a result of ascending infection, most commonly by E. Coli
Presents with fever, chills, sweats, flank pain, renal angle tenderness and leukocytosis +/- frequency, dysuria, haematuria
Leukocytic casts seen in the urine
What is chronic pyelonephritis and reflux nephropathy?
Chronic inflammation and scarring of the renal parenchyma caused by recurrent and persistent bacterial infection
Can be due to:
Chronic obstruciton: posterior urethral valves, renal calculi
Urine reflux
What is acute interstitital nephritis
A hypersensitivity reaction usually to a drug (NSAID, Abx, diuretics)
Usually begins days after drug exposure
Presents with fever, skin rash, haematuria, proteinuria, eosinophilia
What is chornic interstitial nephirits?
Seen in elderly with long term analgesic consumption e.g. NSAIDs, paracetamol
Symptoms occur late in disease: HTN, anaemia, proteinuria and haematuria
What are the thrombotic microangiopathies?
HUS and TTP
What characterises the thrombotic microangiopathies?
Thrombosis generally renal in HUS and widespread in TTP
Triad of:
Microangiopathic haemolytic anaemia
Thrombocytopenia
Sometimes renal failure (HUS)
Pathogenesis of the thrombotic microangiopathies
Widespread fibrin deposition in vessels forming platelet fbirin thrombi, damages passing platelets and RBCs
Leads to platetel and RBC destruction
Resulting in thrombocytopenia and MAHA
HUS
Epidemiology
Pathophysiology
Sign/symptoms
Renal involvement
Dx
Usually affects children
Associated with dairrhoea caused by E. Coli 0157:H7
Can be non-diarrhoea associated due to abnromal protines in the complement pathway
Thrombi confined to kidney
Decreased platelet count-> bleeding, haematemesis, melena
MAHA-> pallor and jaundice
Usually involves renal failure
Dx:
Anaemia, thrombocytopaenia
Signs of haemolyiss: raised bilirubin, LDH and reticulocytes
Fragmented RBCs on blood smear
Coomb’s negative as not AIHA
TTP
Epidemiology
Pathophysiology
Sign/symptoms
Renal involvement
Dx
Usually affects adults
Thrombi occurs throughout circulation with CNS involvement particularlr
Reduced platelets-> bleeding
MAHA-> pallor and jaundice
Usually no renal involvement, neuro symptoms predominate (headache, altered consciousness, seizures, coma)
Dx:
As for HUS
What is ARF?
A rapid loss of renal funciton manifesting as increased serum creatinine and urea
Complications include metabolic acidosis hyperkalaemia, fluid overload, HTN, hypocalcaemia, uraemia
Causes of acute renal failure
Pre-renal (Most cmmon): renal hypoperfusion
e.g. hypovolaemia, sepsis, burns, acute pancreatitis and RAS
Renal:
ATN commonest renal cause of ARF
Acute GN
Thrombotic microangiopathy
Post-renal: obstruction to urine flow
Stones, tumour, prostatic hypertrophy and retroperitoneal fibrosis
Def: Chronic renal failure
Progressive irreversible loss of renal function characterised by prolonged symptoms and signs of uraemia
What are the signs of uraemia?
Fatigue, itching, anorexia, eventually confusion
What are the most common causes of CRF in the UK?
DM (20%)
GN (15%)
HTN and vascular disease (15%)
Reflux nephropathy (10%)
PKD (10%)
With what is HUS associated
E Coli O157: H1
How is CRF classified?
5 stages based on GFR:
Stage 1: kidney damage with normal renal function (proteinuria)
>90
Stage 2: mildly impaired
60-89
Stage 3: moderately impaired
30-59
Stage 4: severely impaired
15-29
Stage 5: renal failure (RRT required)
<15 or if being treated with RRT
Features of Adult PKD
Autosomal dominant inheritance, 85% due to mutations in PKD1 remained in PKD2, both encode polycystin
Pathological features: large multicystic kidneys with destroyed renal parenchyma, liver cysts (PKD1) and Berry aneurysms.
Clinical features: haematuria, flank pain, UTI. Clinical features are often due to cysts cxs e.g. rupture, infection, haemorrhage
Features of lupus nephritis
Depends on site and intensity of immune complex deposition
Presentation may be with
Acture renal failure
Urinary abnromalities
Nephrootic syndrome
or progressive CRF
What is Class 1 Lupus Nephritis
Minimal mesangial lupus nephirits
Immune complexes with no structural alteration
What is Class 2 LN
Mesangial proliferative LN: immune complexes and mild/moderate increase in mesangial matrix and cellularity
What is Class 3 LN?
Focal lupus nephritis: active swelling and proliferation in less than half the glomeruli
What is Class 4 LN?
Diffuse LN involving more than half of the glomeruli
What is Class V LN?
Membarnous LN with subiepithalil complex deposition?
What is Class VI LN?
Advanced sclerosing: complete sclerosis of >90% of the glomeruli
How do NSAIDs predispose to acute tubular injury?
Through inhibition of prostaglandins
Struture of the normal oesophagus
Proximal: squamous epithelium
Distal: columnar (about 1.5-2cm below the diaphragm)
LOS: 2-cm segment proximal to the OGJ
OGJ: point where tubular oesophagus joins saccular stomach
Squamo-columnar junction/ Z line: irregular serrated margin usually at +/- 40cm from incisors but may lie anywhere within distal 2cm
Where is teh squamocolumnar junction?
+/- 40cm from the incisors in distal 2cm of oesoophagus
What is the most common cause of oesophagitis?
What is this?
What is the pathology?
GORD
Reflux of acidic gastric contents into the oesophagus
Ulceration, necrotic slough, inflammatory exudates, granulation tissue, fibrosis
What are the Cxs of GORD?
Haemorrhage, perforation, stricture, Barret’s oesophagus
How is GORD classified?
What does this consitute?
Los Angeles Classificaiton
Grade A: mucosal breaks confined to the mucosal fold, each no longer than 5mm
Grade B: at least one mucosal break longer than 5mm coninfed to the mucosal fold but not continuous between two folds
Grade C: mucosal breaks that are continuous between the tops of the mucosal folds but not circumferential
Grade D: extensive mucosal breaks engaging at least 75% of the oesophageal circumference
What is Barret’s oesophagus?
Re-epithelialisation by metaplastic olumner epithelium with goblet cells
Barret’s: columnar lined oesophagus
Goblet cell/intestinal metaplasia
Precancerous form: metaplastic galndular epithelium-> dysplasia-> Adenocarcinoma
Surveillance: repeat endoscopy and biopsy to detect early neoplastic changes
Adenocarcinoma of the oesophagus
30-40% of primary oesophageal carcinoma
Associated with Barrett’s
Other Cas: squamous or combined form
Features of squamous cell carcinoma of the oesophagus
Dysphagia, anorexia, weight loss
90% in the mid and lower oesophagus
Invasion into the muscularis propria
30-40% have invasion of the mediastinum, 50% have LN mets
Cytology + biopsy
Can spread directly, by LNs or liver mets
Features of oesophageal varices
Extremely dilated submucosal veins in the lower third othe oesophagus.
A consequence of portal HTN commonly due to cirrhosis
Featuires of the normal stomach
Made of the cardia, body and antrum
Lined by gastric mucosa: columnar epithelium (mucin secreting) and glands, then lamina propria and muscularis mucosa
Features of acute gastritis
Acute insult
Neutrophil infiltration
Caused by chemicals e.g. Aspirin/NSAIDs, ETOH or corrosives or infection (H. Pylori)
Features of chronic gastritis
Persistent insult
H. pylori associated
Lymphocytic infiltration
May also by neutrophils and MALT induction. Also associated with chemical, autoimmune and others
Features of chemical gastritis
Reactive or reflux, caused by NSAIDs.
Pattern: foveolar hyperplasia (mucus producing cells), smp and sparse chronic inflammation +/- neutrophils
What differentiates between actue/chronic gastritis
Acute: neutrophil infiltration
Chronic: lymphocyte
Features of helicobacter associated gastritis
H. pylori or Heilmanni
Chronic gastritis +/- activity, severity varias
Outcome variable:
Persistence
Intestinal metaplasia
Dysplasia
Ca and lymphoma
Eradication willl reduce the risk of Ca but will not eliminate cancer due to [redetermined pathways
Causes of pernicious anaemia
Parietal cell Abs (90%)
IF Abs (60%)
Chronic gastritis and body atrophy
Outcome variable: Vit B12, atrophy and Ca
What are some other causes of gastritis
Infection: CMV, Herpes, Strongyloides (immunosuppresion)
IBD: Crohn’s
Why should we treat gastritis?
Chronic gastritis/ulcer
Intestinal metaplasia
Dysplasia
Cancer
Why should alll ulcers be biopsied?
Cxs?
To exclude malignancy
Bleeding (anaemia), perforation, peritonitis
What is intestinal metaplasia a response to?
What is a consideration?
IM in gastric mucosa is a response to LT damage e.g. H. Pylori and Bile
There is a Ca risk
What is gastric dysplasia?
An abnormal pattern of growth in which some of the histological features of malignancy are present but a non or pre-invasive stage
Features of Gastric cancer
High incidence in Japan, Chile, Italy, CHina, Portual and Russia
M>F
90% are carcinomas
Environmental factors: smoking, diet
Mx of duodenitis?
Do a gastric Bx to assess H. pylori status, the principal cause is acid in the presence of gastric metaplasia
Normal architecture of the duodenum?
Villus:crypt 2:1
No increase in lamina propria cellularity
No evdience of epithelial damage, no neutrophils
Brunner’s glands in 1st part
Pathogens in the duodenum?
Immunosuppressed
CMV: microsporidois, crytposporidiosis in immunosuppression
Giarda Lambila Infection
Whipples disease: Tropheryma Whippeli
Endoscopic findings for partial villous atrophy
Show scalloping with a smooth shiny mucosa (or normal)
Bx: early changes are hard to see on biopsy.
There is normal variation in villous height
May be crypt hyperplasia or intraepithelial lymphocytes
Cause of gastric lymphoma
Small intestine?
H. pylori
Coeliacs: 10% will get primary lymphoma (less often carcinoma of the gut) if not adeuqately treated.
Mx of GORD
lifestyle changes (smoking, weight loss), PPI/H2R antag
Prevalence of Barrett’s?
Pathogenesis
Seen in 10% of those with symptomatic GORD
Upwards migration of the SCJ
Where is oesophageaul adenocarcinoma seen?
Lower 1/3rd of oesophagus due to association with Barrett’s
Risk factors for SCC oesophageal carcinoma?
ETOH, smoking
Achalasia of cardia
Plummer-Vinson
Nutritional deficiencies
Nitrosamines
HPV
Presentation of SCC oesophagus
Progressive dysphagia, odynopgagia, anorexia, severe weight loss
Mx of varices
Emergency endoscopy-> sclerotherapy/banding
Def: gastric ulcer
Breach through muscularis mucosa into submucosa
Epigastric pain +/- weight loss
What differentiates between gastric ulcer vs dudodenal ulcer?
Gastric ulcer is WORSE with food
Duodenal ulcer is RELIEVED by food
Ix of gastric ulcer
Biopsy for H. pylori status: punched outm lesion with rolled margins
Rx for H. pylori
Triple therapy
PPI
Clarithromycin
Amoxicillin or metronidazole
Features of coeliac
T-cell mediated autoimmune disease: DQ2, DQ8 HLA status
Villous atrophy and malabsorption
Presents in young children and Irish women (EMQs)
Symptoms of coeliac
Malabsorption: steatorrhoea, abdo pain, N+V, weight loss, fatigue, IDA, failure to thrive, rash (dermatitis herpetiformis)
Serological tests for Coeliac
Anti-endomysial Ab (best sensitivity and specificity)
Anti-TTG
Anti-gliadin (poor marker of disease control)
Gold standard Ix in coeliac?
Upper GI endoscopy and duodenal biopsy
(villous atrophy, crypt hyperplasia, lymphocyte infiltrate
10% progress to duodenal T-cell lymphoma if not adequately treated

Stomach (body)
lined by gastric mucosa columnar epithelium (foveolar, mucin secreting)
specialised glands in the lamina propria
muscularis mucosa

Stomach (antrum)
lined by gastric mucosa columnar epithelium (fovelolar, mucin secreting)
Non-specialised glands in the lamina propria
(gastric pits)
mucularis mucosa

Duodenum
Glandular epithelium
with goblet cells
(intestinal type epithelium)
Villous architecture
villous:crypt ratio of >2:1
Differences between metaplasia, dysplasia, cancer
Metaplastic glandular epithelium
(intestinal type)
Dysplasia changes showing some of the cytological and histological features of malignancy but no invasion through the basement membrane
Adenocarcinoma invasion through the basement membrane
What are the two morphological classifications of gastric cancer?
Intestinal: well differentaited
Diffuse: poorly differentiated Linitis plastica, includes signet ring cell caricnoma

VIllous atrophy
Coeliac disease

Duodenal MALToma
Layers of the skin
Epidermis:
tratum corneum
Stratum lucidum
Stratum granulosum
Stratum spinosum
Stratum Basale
DEF
Dermis:
Paipllary Dermis
Reticular dermis
Subutis
What are these structures?


What is the organisation of the epidermis
Corneum
Lucidum
Granulosum
Basale
DEJ
What are the layers of the skin from superficial to deep?
Horny layer
Granular layer
Squamous cell layer
Basal layer
Epidermis cell activitiy from dep to superfifical
Basalae: mitosis, cells bound to BM by hemidesomosomes
Spinosum (prickle layer): cells linked by desmosomes
S granulosum: nuceli disintegrate
Corneum: non-nucleated, contains keratin


What does hyperkeratosis mean?
Increase in S. corneum/ kertain
What is parakeratosis?
Nuclei in S corneum
What is acanthosis?
Increases in spinosum
What is acantholysis?
Decreased cohesions between keratinocytes
What is spongiosis?
Intercellular oedema
What is legtiginious epidermis?
Linear pattern of melanocyte proliferation within epidermal basa cell layer
Features of dermatitis
All have same histpoapthology
Spongiosis of the epidermis, perivascular chronic inflammatory infiltrate in the dermis
Dilated dermal capillaries
Chronic: acanthosis, crusting/scaling
What are the different types of dermetitis
Atopic (eczma)
Contact
Seborrhoeic
Features of atopic dermatitis
Infants: face, scalp
Older: flexural areas
If chronic may lead to lichenification, persists into adulthood in those with FHx of atopy
Features of contact dermatitis
Type IV hypersensitivity e.g. to Nickel, rubber
Erythema, swelling, pruritus
Commonly affects ear lobes and neck (from jewellery), wrst (leather watch straps), feet (from shoes)
Features of seborrhoeic dermatitis
Inflammatory reaction to a yeast- Malassezia
Infants: cradle cap
Young adults: mild erythema, fine scaling, mildly pruritic affecting face, eyebrow, eyelid, anterior chest, external ear


Features of Lichen Planus
Lesions are pruritic, purple, polygonal, papules and plaques
Mother of pearl sheen and fine network on surface called Wickam’s striae
Usually on inner surfaces of wrists, can also affect oral mucous membranes.
Aetiology unknown
Hyperkeratosis with saw toothing of rete ridges and basal cell degeneration with chornic inflammatory infiltrate
Hyperkeratosis with saw toothing of rete ridges and basal cell degeneration with chornic inflammatory infiltrate
Lichen planus
Wickam’s striae seen in?
Lichen planus
Features of Psoriasis
2%
Commonest form is plaque is chronic plaque psoriasis with salmon pink plaques and a silver scale
Rubbing them causing pin-point bleeding
Koebner phenomenon: lesions form at sites of trauma
Cells have increased proliferation rate
Parakeratosis, loss of granular layer, clubbing of rete ridges giving test tubes in a rack appearance. Munro’s microabscesses
Parakeratosis, loss of granular layer, clubbing of rete ridges giving test tubes in a rack appearance. Munro’s microabscesses
Plaque psoriasis
What is Auspitz’s sign
Rubbing of psoriatic plaques causing pin-point bleeding
What is Koebner phenomenon
Psoriatic plaques forming at trauma sites
What are the types of psoriasis
Chronic plaque psoriasis
Flexural psoriasis: seen in later lift, usually groin, natal cleft and sub mammary areas
Guttate psoriasis: rain drop plaque distribution seen 2 week post Strep-throat
Erthrodermic/pustular psoriasis: severe widespread disease often systemic symptoms
With what is psoriasis associated?
Pitting
Onycholysis
Subungual hyperkeratosis
Arthritis




Features of pityrisasis Rosea
Salmon pink scaly eruption on the trunk extending outwards
Herald patch
May be assocaited with a virus
Pathology: non specific dermatitis
Features of erythema multiforme
Causes annular target lesions on hands and feet.
Pleomorphic lesions that can be a combination of macules, papules, urticarial weals, vesicles, bullae and petechiae
Subepidermal bulllae on histology
Causes of erythema multiforme
Infections e.g. HsV, mycoplasma or drug reactions e.g. penicllin, salicylates, anti-malarials
Spectrum of erythema multiforme
SJS, TEN
Bullous disesease
Def:
Site
Vesicles <0,5cm
Bullae >0.5cm
Can be sub-epidermal, intra-epidermal and subcorneal
What are the three types of bullous disease
Dermatitis herpetiformis
Pemphigoid
Pemphigus
Features of dermatitis herpetiformis
Itchy vesicles on extensor surfaces of elbows, buttocks.
Associated with coeliac
IgA Abs bind to the BM leading to subepidermal bulla
Microabscesses which coalesce to form subepidermal bullae. Neutrophil and IgA deposts at the tips of the dermal papillae
Microabscesses which coalesce to form subepidermal bullae. Neutrophil and IgA deposts at the tips of the dermal papillae
Dermatitis herpetiformis

Dermatitis herpetiformis
Features of pemphiogid
Large tense bullae on erythematous ase. Often on forearms, groin and axillae, seen in the elderly
Bullae do not rupture as early as pemphigus
IgG Abs bind to demidesomosomes of BM-> subepidermal bullae
PemphigoiD: Bullae are deep
Subepidermal bullae with eosinophils.
Linear deposition of IgG along BM
Subepidermal bullae with eosinophils.
Linear deposition of IgG along BM
Pemphiogid
Deep: subepidermal bulla
Features of Pemphigus
Bullae are easily rupturd and found on skin AND mucous membranes
IgG Abs bind to desmosomal protines-> intraepidermal bulla
PemphiguS- superficial
Intraepidermal bulla. Netlike pattern of IgG deposits
Acantholysis
Intraepidermal bulla. Netlike pattern of IgG deposits
Acantholysis
Pemphigus


Features of lupus rash
Chronic, discoid LE, buttterfly rash, alopecia


What are the types of cutaenous tumours
Benign:
Seborrhoeic keratosis
Premalignant:
Actinic Keratosis
Keratoachthoma
Bowen’s Disease
Malignant:
SCC
BSS
Melanocytic:
Melanocytic naevia
Lentigines
MM
Features of seborrhoeic keratitis
Rough, waxy plaques, stuck on appear in middle age/elderly
Features of Actinic keratosis
(Solar keratosis)
Rough, sandpiper like lesions on sun-exposed areas
SPAIN:
Solar elastosis
Parakeratosis
Atypia/dysplasia
Inflammation
Not full thickness
SPAIN:
Solar elastosis
Parakeratosis
Atypia/dysplasia
Inflammation
Not full thickness
Actinic keratosis
Features of keratoacanthoma
Rpaidly growing dome shaped nodule which may develop a necrotic crusted centre.
Grows over 2-3w and clears spontaenously
May be difficult to differentiate from SCC hsitologically
Features of Bowen’s disease
Intra-epidermal SCC in situ
Flat, red, scaly patches on sun-exposed areas
Full thickness atypia/dysplasia with BM intact
Full thickness atypia/dysplasia with BM intact
Bowen’s disease
Features of SCC
When Bowen’s has spread to involve dermis. Similar clinical features to Bowen’s but may ulcerate
Atypia/dysplasia throughout epdiermis, nuclear crowding and spreading through BM into dermis
Atypia/dysplasia throughout epdiermis, nuclear crowding and spreading through BM into dermis
SCC
Features of BCC
Aka: rodent ulcer
Slowly growing tumour, rarley metastatic but locally destructive
Pearly surfact often with telangectasia
Mass of basal cells pushing down into dermis. Nuclei align in outermost layer (pallisading)
Mass of basal cells pushing down into dermis. Nuclei align in outermost layer (pallisading)
BCC
Features of melanocytic naevi
Proliferation in the basal layer, nesting into dermis
What are lentigines?
Melanocyte proliferation limited to the epidermis
Features of MM
Atypical melanocytes which intiially grow horizontally in the epidermis (radial growth phase), then grow vertically into dermis (vertical growth phase). Vertical growth produces Buckshot appearance (=pagetoid cells)
What is the most important prognostic factor for MM?
Breslow Thickness
ABCD of MM
Asymmetry
Border irregularity
Colour
Diamete >6mm
What are the different classifications of MM
Lentigo MM
Superficial spreading MM
Nodular MM
Acral lentiginous MM
Features of lentigo MM
Sun exposed areas of elderly caucasians: flat, flowly growing black lesions. MM can develop in a lentigo MM
Features of superficial spreding MM
Irregular borders with variation in colour. Not associated with any special site
Features of nodular MM
Can occur at all sites and seen in younger age group
Features of acral lentiginous MM
Palms, soles and subungual areas.

Erythema multiforme








SJS/TEN features
Dermatological emergency: sheets of skin detachment
Nikolsky sign positive: mucosal involvement prominent
Commonly caused by drug e.g. sulfonamide antibiotics
SJS vs TEN
<10% body surface vs >30%
Features of skin appenage tumours
Hair follicles and seat glands can produce specialised tumours of skin
What are the different types of cutaneous vascular tumours
Capillary haemangioma- strawberry mark
Flat haemangioma- port wine stain
Kaposi’s sarcoma
What is mycosis fungiodes?
Most common form of cutaenous T-cell lymphoma generally affects skin.
Is a NHL
Has a muschroom like appearance
Hisotlogical features of herpes infection of skin
Cluster of inflamed pustules and vesicles



Viral infection




Pemphigoid

Pemphigus
Function of cortical bone
Aka Compact bone
Facilitaties mane bone functions: suppors, protection and elverage
Forms cortex, dense hard bone to which muscles are attached
Function of trabecula bone
Also called cancellous bone
Higher SA, less dense, softer, weaker and less stiff,
High vascular and contains red bone marrow where haematopoesis.
Vertebrae
Has role in homeostasis of Ca
What are the types of non-neiplastic bone disease
Trauma
Infeciton
Degeneration
Inflammation
Metabolic
What are the classifications of fractures
Complete or incomplete
Closed
Comminuted
Compound


What are the stages of # repair
Organisation of a haematoma at # site (pro-callus)
Formation of fibrocartilaginous callus
Mineralisation of fiberocartilaginous callus
Remodelling of bone along weight bearing lines
What are the factors influencing #] repair
Type
Presence of infection
Pre-existing conditions e.g. neoplasm, metabolic, drugs (steroids), Vit deficiency
What are the features of osteomyelitis in adults
Sites: direct spread, secondary to diabetic skin ulcer, dental abscess etc.
S. aureus (90%)
E coli
Kleb
Salmonella (associated with SCD)
Pseudomonas IVDU
What are the features of osteomyelitis in children
Haematgoenous spread to long bones, usually metaphytic
Neonates: Haemophils influenza, Group B strep
Clinical features of osteomyelitis
Fever, malaise, chills, leucocytosis
Local: pain, swelling and tenderness
Blood test +ve in 60%
XR: mixed picture eventually lytic, 10d post onset
Periosteal reaction
Mottled rarefaction (increased osseus vascularity and lifting up of the periosteum)
Involcrum: after 1w irregular sub-periosteal bone formation
Lytic destruction (10-14d)
Seqeustra: after 3-6w, some areas of cortex may become detached
XR features of osteomyelitis
Periosteal reaction
Involcrum (after 1w: sub-periosteal bonefrmation)
Irregular lytic destruction
Sequestra: 3-6w, necrotic areas of cortex may become detached.
What is the clinical staging of osteomyelitis
Cierny-Mader Staging
Anatomical Type: 1- medullary, 2- superficial, 3- localised, 4- diffuse
Physiologic: Host A-normal, B- local or systemic compromise, C- treatment worse than disease
Mx of osteomyelitis
Biopsy: culture
6w IV antibiotics
Features of TB osteomyelitis
Rare cause mainly affecting immunocompromised
More destructive and resistant to contral
Spinal disease may result in psoas abscess: inguinal lump and severe skeletal deformity: Pott’s
Systemic amyloidosis may result in protracted cases as an inflammatory response to chornic TB infection
Marrow replaced by inflammatory response and a granuloma forms. There are Langerhan’s type giant cells which have a peripheral rim of nuceli
Marrow replaced by inflammatory response and a granuloma forms. There are Langerhan’s type giant cells which have a peripheral rim of nuceli
TB osteomyelitis
With what are Langerhan’s type giant cells associated
TB
Features of Syphillitic OM
Caused by Traponema pallidum
Can be congenital or acquired
Congenital: skeletal lesions: osteochondritis, osteoperiostitis, diaphyseal ostomyelitis
Acquired: sekeltal lesions are late onset, require lack of Rx:
non gummatous periosteitis
gummatous inflammation of bone and joints
neuropathic joints (tabes dorsalis)
neuropathic shaft fractures
non gummatous periosteitis
gummatous inflammation of bone and joints
neuropathic joints (tabes dorsalis)
neuropathic shaft fractures
Syphillitic OM
Features of Lyme disease
Inflammatory arthropathy as part of a complex multisystem illness resulting from tick bite
Caused by Borrelia burgdorferi: ixodes dammini (tick spp.)
Can be mistaken for OM
Associated with erythema chronicam migrans
What are the stages of Lyme disease?
1: early localisation, selling + rash (90%) usually within 7-10d and in groin, axilla, earlobe or on thigh
Stage 2: early dissemination, affects multiple organ systems
Stage 3: low grade, late and persistent, dominated by arthritis, mimics RA
Mx of Lyme disease
Prevention: long trousers, vaccines
Antibiotics for proven disease
Dx is purely clinical

OM
What is a Brodie abscess?
Subacute OM
Features of OA
Degenerative joint disease:
Disease of cartilage, cartialge is lost and osteophytes formed, pain due to friction
Eboination of underlying bone
Avascular necrosis of the femoral head can occur
Cartilage degernation
Fissuring
Abnormal matrix calcification
Osteophytes
What is the difference between primary and secondary OM
Primary is age related
Secondary is to a prveiously damaged joint or congenitally abnormal joint
XR changes in osteoarthritis
Loss of joint space
Osteophytes
Subchondral sclerosis
Subchondral cysts
Clinical features of OA
Big weight bearing joints, vertebrae and kness
PIPJ: Bouchards’
DIPJ: Herbeden
May also affect MCP
Synovial disease: synovium Bx is non-specific with increased vessels and chronic inflammation
XR features of RA
Loss of joint space
Erosions
Soft Bones
Soft tissue swelling
Features of RA
Disease of the synovium/severe chronic relapsing synovitis
Slow and progressive.
F>M
RF +ve in 80%, immunocomplexes are more associated with extra-articular disease
What factors are involved in genetic predisposition to RA
HLA-DR4 and DR1
TNF, STAT
Clinical features of RA
Hand: radial deviation of wrist and ulnar deviation of fingers
Swan neck deformity: extension of POPJ and flexion of DIPJ
Boutonniere: flexion PIPJ
Z shaped thumb
Symmetrical pain and swelling
DIPJ swelling
Small joints of hands and feet, wrists, elbows, ankles and knees
Mild anaemia, raised ESR, RF +ve, rheumatoid nodules?
Swan neck deformity
Extension of PIPJ and flexion of DIPJ
Boutonniere deformity
Flexion PIPJ, extension of DIPJ
Histopathology of RA
Proliferative synovitis with thickening of the synovial membranes, hyperplasia of surface synoviocytes, intense inflammatory cell infiltrate, fibrin deposition on the joint and encrosis
Pannus: exuberant inflamed synovium of the articular surface, hyperplastic synovium.
Proliferative synovitis
Grimley-Sokoloff cells
What are Grimley-Sokoloff cells
Hyperplastic synovial cells
What are the stages of RA
Unknown antigen reaches the synovial membrane
T-cell proliferation associated with increased B cells and angiogenesis
Chronic inflammation with inflammatory cytokines
Pannus
Cartilage and bone destruction
What differentiates between RA and OA in hands
RA spares DIPJ

RA

OA
Features of Gout
Male, middle aged
Increased dietary purin intake, ETOH, diuretics, inherited metabolic abnormalities
Great toe: MTP (podagara), lower extremities
Urate crystals: needle shaped
Negatively birefringent crystals
Clinical features of gout
Hot, swollen, red, painful joint.
Tophus is the pathognomic lesion (on pinna and hands)
Negatively birefringent crystals=
Urate
GOUT
Featurs of pseduogout
>50y
Idiopathic, hyperPTH, DM, hypothyroid, Wilsons
Knee and shoulder
Hot, swollen joint with effucion
Calcium pyrophosphate crystals, rhomboid shaped
Positively birefringent
Positively birefringent crystals=
Pseudogout
Mx gout
Acute attack: colchicine
LT: allopurinol
Conservative: ETOH and purine intake (sardines/liver) reduction
Mx pseudogout
NSAIDs or intra-articular steroids
Subtypes of pseudogout
Sporadic
Metabolic: haemachromatosis, primary HPT, mypo Mg, low PO4
Hydroxylapatite crystal deposition
Hereditary: AD, ANKH mutation, transmembrane protein
Traumatic: e.g. bad OA of the knee
Crystals in pseudogout
Calcium pyrophosphate (mainly knees) or calcium hydroxyapatite (knees and shoulders)
Crystals survive processing, whereas crystals of gout dissolve in aqueous solution and need to be fixed in alcohol or sent unfixed.
Crystals are not needle shaped but found in polyhonal or rhomboid islands

Gout

TB OM

AVN of femoral head with characteristic wedge shaped infarct

Swan neck deformity

Boutonniere

RA vs OA synovium
RA= proliferative
What is the most common site for bone malignancy?
Knee
What is the clinical presentation of bone tumours?
Pain, swelling, deformity and fracture
Diagnosis of bone tumours
XR: evaluate site, margin and size. Number of lesions and soft tisse extension. Any associated disease or fratcure
Biopsy: performed by radiologist with a Jamshidi needle to produce a core lesion. Can be CT or USS duiged. Open Bx for sclerot or inaccessible lesions
What are the different types of malignant bone tumour?
Osteosarcoma
Chondrosarcoma
Eqing’s sarcoma
Giant cell (borderline malignancy)
What are the most common malignant bone tumours?
Secondary metastatic
What adult malignancies metastasise to bone?
Breast
Prostate
Lung
Kidney Thyroid
What childhood malignanices commonly metastasise to bone?
Neuroblastoma, Wilm’s, osteosarcoma, Ewings, rhabdomyosarcoma
Features of osteosarcoma
Adolescents. M>F. 75% 10-30y.
Site: knee>tibia>humerus>skull>femur
Originates from cells of mesenchymal origin, osteoblastic differentiation differentiation and produce malignant osteoid
Commonest primary bone tumour
Malignant mesenchymal cells +/- bone and cartilage formation. H&E stain of osteosarcoma will show mixed malignant bone in a backgrown of less cellular chondroid tumour. ALP +VE
Malignant mesenchymal cells +/- bone and cartilage formation. H&E stain of osteosarcoma will show mixed malignant bone in a backgrown of less cellular chondroid tumour. ALP +VE`
Osteosarcoma

osteosarcoma

Osteosarcoma
ALP +ve
XR appearance of osteosarcoma
Usually metaphyseal
Lytic
Permeative
Elevated periosetum (Codman’s triangle)
Classification of osteosarcoma
Site within bone: intramedullary, cortical or surface
Degree of differentiation: grade
Multicentricity: synchronous or metachronous
Primary or secondary
Mx of osteosarcoma
CTx and limb salvage surgery
Features of chondrosarcoma
Malignant cartilage producing sarcoma
>40
M>F
Pelvis, axial skeleton, proximul femur, proximal tibia
XR: ltyic lesions with fluffy calcification involving axial skelton
Malignant chondrocytes +/- chondroid matrix may dedifferentiate to high grade sarcoma
Px: 70% 5y but depends on grade and size
XR: ltyic lesions with fluffy calcification involving axial skelton
Chondrosarcoma
Malignant chondrocytes +/- chondroid matrix may dedifferentiate to high grade sarcoma
Chondrosarcoma
Classiicaiton of chondrosarcoma
Site: intramedullary or juxtacortical
Histologically: conventional (myxoid or hyaline), clear cell, dedifferentiated or mesenchymal
Features of Ewing’s Sarcoma
AKA PNET (primitive peripheral neuroectodermal tumour)
Highly malignant small round cell tumour found in bone and soft tissue
<20y/0
Long bones and pelvis: diaphysis or metaphsis
XR: Onion skinning of periosteum
Sheets of small round cells. CD99+ve, T 11.22 translocation
Px: 75% 5y
XR: Onion skinning of periosteum
Ewing’s Sarcoma
Sheets of small round cells. CD99+ve, T 11.22 translocation
Ewing’s Sarcoma

Ewing’s Sarcoma
Onion skinning of the periosteum
With what is chromosomal translocation of 11:22 associated?
Ewing Sarcoma
What are the different types of Round cell tumours?
Ewing
Small cell osteosarcoma
Rhabdomyosarcoma
Neuroblastoma
Lymphoma
SCC
What are the features of Giant Cell Tumours
Borderline malignancy
20-40y
F>M
Epiphysis with metaphyseal extension
Knee epiphysis, either femur or tibia, wrist, pelvis, sarcum
XR: lytic lesions right up to articular surface
Osteoclast-type multinucleate giant cells on background of spindle/ovoid cells
Usually benign with unpredicatble behaviour
Locally aggreswsive, may recur and can metastasise
XR: lytic lesions right up to articular surface
Osteoclast-type multinucleate giant cells on background of spindle/ovoid cells
Giant Cell Tumour

Giant cell tumour
Lytic lesion right up to articular surface
Features of malignant bone tumours
60 times rarer than lung Ca
Primary malignant bone tumours are most cmmon in children and young adults
On H&E appear red and destroy local bone
What are the types of benign bone tumours
Osteid osteoma
Osteoma
Enchondroma
Osteochondroma
Fibrous dysplasia
Simple bone cysts
Osteoblastoma
What are the features of fibrous dysplasia
Normal bone replaced with fibrous bone tissue causing abnromal sweeling and growth of bone.
F>M, after 3rd decade
Any bone: ribs and proximal femur commonest. Monosototic>polyostotic
XR: soap bubble osteolysis. Shepher’d crook deformity
Irregular trabeculae of woven bone in bland spindle cell stroma. Bone trabeculae shapes= chinese letters
XR: soap bubble osteolysis. Shepher’d crook deformity
Irregular trabeculae of woven bone in bland spindle cell stroma. Bone trabeculae shapes= chinese letters
Fibrous dysplasia

Shepherds Crook
Fibrous dysplasia

Chinese letters
Fibrous Dysplasia
Albright syndrome
Polyostotic fibrous dysplasia
Cafe au lait spots
Precocious puberty (endocrine problems)
What are the tumour like coniditions of bone?
Fibrous dysplasia
Metaphyseal fibrous cortical defect/non-ossifying fibroma
Reparative giant cell granuloma
Ossifying fibrome
Simple bone cyst
What are the features of simple bone cysts?
Found in humer or femur
Fluid filled and unilocular, lystic well defined margins
How can the benign tumours of bone be classified?
Cartilaginous differentiation:
Osteochondroma
Endochrondroma
Chondroblastoma
Bone forming tumours:
Osteoid osteoma
Osteoblastoma
Osteoma
What are the features of osteoid osteomas
Adolescent
M>F
Arise from osteoblasts, tend to be <1.5cm, most common in long bones (tibia and femur)
Night pain relieved by aspirin
Radiolucent nidus with sclerotic rim
Normal bone
Radiolucent nidus with sclerotic rim
Normal bone
Osteoid osteoma
What are the features of osteoma
Middle age
A new piece of bone usually growing on another piece of bone, tpically inthe head and neck
Normal bone
Assocaited with Gardner syndrome
What is Gardner Syndrome?
GI polyps + multiple osteomas + epidermoid cysts
What are the features of osteoblastomas?
Similar to osteoid osteomas. Osteoblastoma is a giant osteoid osteoma
XR: speckled mineralisation
XR: speckled mineralisation
Osteoblastoma

Osteoid osteoma
Radiolucent nidus with sclerotic rim

Osteoblastoma
Speckled mineralisation
What are the features of enchondromas?
Benign tumours of cartilage.
55% of pts 10-40y
Hand>humerus>knee>foot
XR: lytic lesion. Cotton wool calcifcation. Expansile O-ring sign
XR: lytic lesion. Cotton wool calcifcation. Expansile O-ring sign
Enchondroma
Features of osteochondroma
50%pts 10-20y. M:F
Knee, humerus, tibia and femur. Growth plates of long bones
A benign cartilage capped bony outgrowth.
XR: well defined bony protruberance from bone
Cartilage capped
Most common neoplsam of the skeletonl.
If there is irritation it can be removed by surgery
XR: well defined bony protruberance from bone
Cartilage capped
Osteochondroma
Features of chondroblastomas
Rare, originating from chondroblasts, most common epiphyseal tumour in children
Ollier’s Syndrome
Multiple endochondromas
Maffuci’s syndrome
Multiple endochondromas with haemangioma
Diaphyseal aclasis/ hereditary multiple exostoses
Multiple exostoses (osteochondromas) + short stature + bone deformity
What is the definition of soft tissue tumour
Mesenchymal proliferation which occur in the extraskeletal non-epithelial tissues of the body excluding the meninges and the lymphoreticular system
Anywhere but the majority occur in the large muscles of extremities, chest wall and retroperitoneum.
Mostly older. M>F but varies by histological type
What are the types of soft tissue sarcoma?
Myxoid liposarcoma
Spindle cell sarcoma
Pleomorphic tumours
Synovial sarcoma: biphasic patter with epithelial and spindle cell areas.
What are the congenital lower GI diseases?
Atresia and stenosis
Duplication
imperforate anus
Hirschprung’s
What are the features of Hirschprungs?
Absence of ganlgion cells in myenteric plexus
Distal colon fails to dilate
80% male
Constipation, abdominal distension, vomiting and overlow diarrhoea in young babies. Associated with DS
Genetics: proto-oncogene Cr10+
Dx and Mx of Hirschprung’s
Clinical impression, biopsy of affected segment: hypertrophied nerve fibres but no ganglia
Mx: resection of affected: constricted segment
How can the acquired diseases of the lower GI be classified?
Mechanical
Inflammatory
Ischaemic
Malignant
What are the causes of GI obstruction
Constipation
Diverticular disease
Adhesions
Herniation
External mass
Volvulus
Intussuception
What is volvulus?
Complete twisting of bowel loop at mesenteric base around a vascular pedicle.
Intestinal obstruction +/- infarciton.
Infants: commonly small bowel
Elderly: sigmoid>caecal
Features of diverticular disease
High incidence in West due to low fibre diet and high intraluminal pressure
Herniation occurs at weak points in the wall of the bowel, 90% in left colon.
Seen as outpouchings on endoscopy.
Cxs: diverticulitis, gross perforation, fistula to bowel, bladder or vagina, or obstruction
What are the inflammatory causes of bowel disease?
Acute:
Infection
Toxins (abx)
CTx/RTx
Chronic:
IBD
TB
Causes of acute colitis
Infection, durgs and toxins, CTx
Features of infective colitis
Secretory diarrhoea (toxin)
Exudative diarrhoea (invasion and muscoal damage)
Severe damage and perforation
Systemic illness
What is pseudomembranous colitis
Abx associated colitis.
Acute colitis with pseudomembrane formation. Caused by protein exotoxins of C. diff
Dx; histology, biopsy, C diff toxin or stool assay
Rx: metronidazole or vancomycin
Features of iscahemic colitis/ infarction
Can be acute or chronic
Most common vascular disorder of the GIT
Usually occurs in “watershed” zones e.g. splenic flexure (SMA/IMA) and the rectosigmoid (IMA/IIA)
Can cause damage in tissue layers leading to perfroation: muscoal, mural, transmural
Causes of iscahemic colitis
Arterial: atheroma, thrombosis and embolism
Venous: thrombus, hypercoagulable states
Small vessel disease: DM, cholesterol emboli, vasculitis
Low flow state: CCF, haemorrhage, shock
Obstruction: hernia, intussuception, volvuluis and adhesions
Clinical features of IBD
Diarrhoea +/- blood
Fever
Abdo pain
Acute abdomen
Weight loss
Extra-intestinal manifestations
Crohn’s epidemiology
Western populations
F>M
White>non-white
Smoking aggravates
UC epidemiology
More common than Crohn’s
Whites>non-whites
20-25y
Aetiology of IBD
Genetic predispotiion (familial aggregation, twin studies, HLA0
Infection (MTB, measles)
Abnormal host immunoreactivity
Pathophysiology of Crohn’s
Affects whole GIT
Most common in terminal ileum and caecam
Skip lesions: patchy distribution
Cobblestone appearance: areas of health mucose lie above diseased mucosa
Apthous ulcer.
Rosethorn ulcers can join together to form serpentine granulomas
Transmural inflammation
Non-caseasting granulomas
Fistula/fissure formation more common
Skip lesions
Crohn’s
Apthous ulcer
First lesion in Crohn’s
Non-caseating granulomas with transmural inflammation
Crohn’s
Pathophysiology of UC
Extends proximally from rectum
Continuous
Small bowel not affected unless v. severe colitis casuses backwash ileitis
Extensive superficial broad ulcers confined to the mucosa
No granulomas/fissures/fistulae/strictures
Islands of regenerating muscoa bulge into lumen to cause pseudopolyps
Mucosal continous ulceration with no granulomas
UC
Backwash ileitis
Severe pancolitic UC
Thick rubber hose like bowel wall with narrow lumen
Fat wrapping
Crohn’s
Bowel wall normal thickness in IBD
UC
IBD:
Intermittent diarrhoea, pain and fever
Crohn’s
IBD:
Bloody diarrhoea, mucus, crampy abdominal pain relieved by defacation
UC
Extra GI manifestation of IBD
Malabsorption and Fe def: stomatitis
Eyes: uveitis, conjuncitvitis
Skin: erythena nodosum, pyoderma gangrenosum, erythema multiforme, digital clubbing
Joints: migratory asymmetrical polyarthropathy of large joints, sacroiliitis, myositis, ankylosing spondylitis
Pericholangitis, primary sclerosing cholangitis, steatosis
PSC
UC
Pancolitis= increased
Extra-intestinal manifestation of IBD:
Eyes
Uveitis, conjunctivitis
Extra-intestinal manifestation of IBD:
Skin
Eythema nodosum
Pyoderma gangrenosum
Erythema multiforme
Clubbing
Extra-intestinal manifestation of IBD:
Joints
Migratory asymmetrical polyarthritis
Sacroilitis
Anky spond
Myositis
Extra-intestinal manifestation of IBD:
Liver
Pericholangitis
PSC
Steatosis
Cxs of Crohn’s
Strictures (recurrent)
Fistulae
Abscess fromation
Perforation
Cxs of UC
Severe haemorrhage
Toxic megacolon
30% require colectomy within 3y for uncontrollable symptoms
Adenocarcinoma (20-30x risk)
Ix in Crohn’s
Markers of inflammation: ESR, CRP, Ba contrast, endoscopy
Ix in UC
Rectal biopsy
Flexible sig/colonoscopy
AXR
Stool culture
Mx Crohn’s
Mild: prednisolone
Severe: IV hydrocortisone, metronidazole
Additional therapies:
Azathioprine, methotrexate, infliximab
Mx UC
Mild: prednisolone and 5 ASA (meslalzine)
Moderate: prednisolone, 5ASA and steroid eneme BD
Severe: admit, nbm IV fluids and IV hydrocortisone, rectal steroids
For remission: 5ASA 1st line, azathioprine 2nd line
Features of carcinoid syndrome
Diverse group of tumours of enterochromaffin cell origin
Produce 5-HT
Commonly found in the bowel but also lung, ovaries, testes, usually slow growing
Features of carcinoid syndorme
Bronchoconstriction
Flushing
Diarrhoea
Features of carcinoid crisis
Life threatening vasodilation, hypotension, tachycardia.
Bronchoconstriction
Hyperglycaemia
Ix of carcinoid syndrome?
Rx
24h urine 5-HIAA- main 5HT metabolite
Octreotide: somatostatin analgoue
What are the non-neoplastic polyps of the large bowel?
Hyperplastic
Inflammatory: pseudopolyps
Hamartomotous (juvenile, Peutz Jeghers)
What are the features of adenoma
Benign dysplastic lesions that are the precursor to most adenocarcinomas
Mostly asymptomatci so need surveillance if >3.4cm
Classified based on architecture as tubular, tubulovillous or villous adenoma
Size is most important risk factor for malignancy, in addition to degree of dysplasia and increased villous component
What can villous adenoma lead to and why?
Hypoproteinaemic hypokalaemia because they leak large amounts of protein and K
What is the sequence of metaplasia in adenocarcinoma
Normal colon-> at risk mutation after first hit mutation in APC
At risk_> adenoma after second hit to APC
Progression to APC follows activation of KRAS, LOF in p53
Features of hamartomatous polyps
Found sporadically in some genetically acquired syndromes
Features of Juvenile polyposis
Focal malformations of mucosa and lamina propria
Generally <5y/o.
Mostly in rectum leading to bleeding
Usually solitary but >100= juvenile polyposis which may require colectomy to stop haemorrhage
Features of Peutz-Jeghers
Multiple polyps, mucocutaneous hyperpigmentation, freckles around mouth, palms and soles
Increased risk of intussucception and malignancy.
Require regular surveillance of GIT, pelvis and gonds
Features of hyperplastic polyps?
Seen at 50-60y and though to be caused by shedding of epithelium-> cell build up
What are the mesenchymal lesions of the GIT?
Stromal lesions
Lipoma
Sarcoma

Villous adenoma

Adenoma

Hyperplastic polyps
Epidemiology of CRC
2nd commonest cause of cancer death in UK
60-79y/o
<50y= ?familial syndrome
98% are adenocarcinomas, 45% in rectum
Aetiology of CRC
Diet low in fibre high in fat
Lack of exercise
Obesity
Familaly syndromes
Chronic IBD
NSAID protective (COX2 overexpressed in 90%)
Clinical features of right sided CRC
Fe def, anaemia. Weight loss
Clinical features of left sided CRC
Change in bowel habit, crampy LLQ pain
Ix in CRC
Sigmoidoscopy, solonoscopy, proctoscopy
Barium enem
Bloods: FBC
CT/MRI
CEA to moniotr disease
How is CRC graded and staged?
Grade: level of differentiation
Staging: Duke’s
Outline Duke’s staging
A: confined to the wall of the bowel >95%
B: Through wall of bowel
1: extending into muscularis propria 67%
2: transmural invasion without LN involvement 54%
C: LN metastases
1: Extending into muscularis propria with LN involvement 43%
2: Transmural with involved LNs 23%
D: Distant mets <10%
Mx of CRC
Sx:
Rectal cancer/ low sigmoid cancer:
<1-2cm above anal sphincter (lower 1/3rd of rectum): abdomino-perineal resection
>1-2cm above anal sphincter: anterior resection
Sigmoid cancer: sigmoid colectomy
Descending colon and distal transverse: left hemicolectomy
Caecum, ascending colon and proximal transverse: right hemicolectomy
Transverse colon: extended right hemicolectomy
RTx to reduce local recurrence post-Sx
CTx in pallliation: 5 fluorouracil
Features of FAP
70% AD mutations in APC gene with 30% AR mutation in MMR genes
Presents 10-15y with >100 adenomatous polyps required for Dx
All will progress to adenoarcinoma if left untreated thereefore most have prophylactic colectomy
Increased risk of neoplasia elsewheree.g. ampulla of Vater and stomach
At birth hypertrophy of retinal pigment epithelium
Gardner Syndrome
Same clinical, pathological and aetiological features as FAP
Distinctive extra-intestinal manifestations:
Multiple osteomas of skull and mandible
Epidermoid cysts
Desmoid tumours
Dental caries
Post-surgical mesenteric fibromatoses
Turcot’s syndrome
Malignant tumours of the CNS associated with FAP
HNPCC
AD mutation in MMR gene
3-5% of colorectal cancer at early age of onset
Cacrinomas usually in right colon, few polyps but fast preogression
Also assocaited with endometrial, ovarian, small bowel, transitional cell and stomach carcinoma
Lynch syndrome
HNPCC
Lynch syndrome I
Familial colon cancer
Lynch syndrome 2
Associated with other cancers of the GIT or reproductive system
What are the Amsterdam criteria
Used to determine testing of individuals at risk of HNPCC
Amsterdam Criteria:
Three or more family members with a confirmed diagnosis of colorectal cancer, one of whom is a first degree (parent, child, sibling) relative of the other two
Two successive affected generations
One or more colon cancers diagnosed under age 50 years
Familial adenomatous polyposis (FAP) has been excluded
Amsterdam Criteria II:
Three or more family members with HNPCC-related cancers, one of whom is a first degree relative of the other two
Two successive affected generations
One or more of the HNPCC-related cancers diagnosed under age 50 years
Familial adenomatous polyposis (FAP) has been excluded
What are the samples in cytopathology?
Gynae: NHSCSP
Non gynae: exfoliative samples
Features of the NHS CP
Women invited from 25-65y
Samples taken are liquid based cytology
Viewed microscopically
Also test for HPV and STDs
Reduces incidence of invasive squamous cell carcinoma
What are the high risk types of HPV
What account for 70% of cervical cancers
16 and 18
What are the significant mutations in lung adenocarcinoma?
EGFR, ALK-1
What are the significant mutations in melanoma
BRAF
What are the significant mutations in breast Ca
BRAC1/2
CERB-B2
What are the significant mutations in CRC
APC
KRAS
Def: DM
Metabolic disorder characterised by chronic hyperglycaemia due to lack of insulin.
Aetiology T1DM
T2DM
Autoimmune destruction of insulin producing beta cells by CD4 and CD8 T lymphocytes
Strongly linked to insulin resistance and obesity
Presentation of DM
Polyuria (osmotic diuresis)
Polydipsia (raised plasma osmolality)
Hyperglycaemia predisposes to recurrent UTI and skin infections
T1 can present as DKA
Dx of DM
Fasting glucose >7mmol/l
Random >11.1mmol/l
Cxs of DM
IHD
CKD
Blindness
PVD
Foot ulceration
Ectopic pancreas
Pancreatic tissue located in the duodenum , stomach or Meckel’s
25-50% asymptomatic in adulthood
Pancreas divisum
Failure of fusion of the ventral and dorsal panceatic duct system
Higher risk of pancreatitis
Annular pancreas
Partial or complete obstructionof the duodenum by the pancres.
Duodenum surrounded by a ring of pancreatic tissue which is continuous with the head of the pancreas
Exocrine functions of the pancreas
Produces 2l/d of enzymic HCO3- rich fluid, stimulated by secretin and CCL
Secretin features
Produced by s-cells of the duodenum
Controls gastric acid secretion and buffering with HCO3
CCK features
Responsible for stimulating digestion of fat and protein
Made by I-cells in the duodenum
Causes release of digestive enzymes
Alpha cells of pancreas
Glucagon-> increases blood glucose
Delta cells of pancreas
Somatostatin to regulate alpha and beta cells
D1 in pancreas
Vasoactive peptide stimulating the secretion of H2O into the pancreatic system
PP in pancreas
Pancreatic polypeptide
Self-regulates secretion activities
Macrovascular cxs of DM
Cardiac- MI
Renal- GN, pyelonephritis
Cerebral- CVA
Microvascular Cxs of DM
Diabetic retinopathy
Claudidcation
Causes of acute pancreatitis
I GET SMASHED
Idiopathic
Gallstones
Ethanol
Trauma
Steroids
Mumps
Autoimmune
Scorpion Venom
Hyperlipidaemia
ERCP
Drugs e.g. thiazides
Presentation of acute pancreatitis
Severe epigastric pain radiating to back, relived by sitting forward.
Vomiting prominent
Hypoetnsive shock in severe cases
A signficantly raised plasma amylase in clinical setting is virutally diagnostic
Can result in the formation of a pseudocyst, associated with alcoholic pancreatitis
What cells perform the exocrine function in the pancreas?
Acinar cells
Amylase vs lipase in pancreatitis
Amylase is only transiently raised
Lipase is more sensitive
Cxs of acute pancreatitis
Severe cases may lead to shock and multiorgan failure
Infection of necrosed pancreas worsens prognosis
Histology in acute pancreatitis
Coagulative necrosis
Causes of chronic pancreatitis
Alcoholism
CF
Hereditary
Pancreatic duct obstruction e.g. stone/tumour
Autoimmune IgG4 sclerosing
IgG4 sclerosing disease
Autoimmune chronic pancreatitis
NB re chronic pancreatitis
Can very closely mimic pancreatic cancer clinically, radiologically and pathologically
What is this tissue?

Pancreas
Presentation of chronic pancreatitis
Chornic upper abdominal pain
Weight loss
Steatorrhoea and diarrhoea occur when most of the gland has been destroyed.
Pathology of Chronic pancreatitis
Very similar to pancreatic carcinoma:
Grossly replaced with fibrous tissue, loss of exocrine tissue, duct dilatation with thick secretions, calcification
What are the features of Acinar cell carcinoma
Rare (1%) malingnant epithelial neiplasm of the pancreas demonstrating evidence of enzyme production by the neoplastic cells
Older adults.
Presentation of acinar cell carcinoma
Non specific symptoms: abdominal pain, weight loss, N+D
10% get multifocal fat necrosis and polyarthralgia due to lipase secretion
Histopathology of pancreatic acinar carcinoma
Neoplastic epithelial cells gowing in sheets
Some have abundnant eosinophilic granular cytoplasm
Positive immunoreactivity for lipase, trypsin, chymotrypsin.
Px of acinar cell carcinoma?
5YSR= <10%
What type of pancreatic malignancy is this?

Acinar cell carcinoma
neoplastic epithelial cells growing in sheets, trabeculae and acini, some have abundant eosinophilic
granular cytoplasm, positive immunoreactivity for lipase, trypsin and chymotrypsin.
What is the most common pancreatic malignancy?
Epidemiology?
Ductal cell adenocarcinoma of the pancreas
M>F
>60y/o
What are the risk factors for pancreatic carcinoma?
Smoking is the main
Diet
Genetic e.g. FAP, HNPCC
What are the clinical features of ductal cell carcinoma
Cachexia and anorexia
Upper abdominal and back pain that is persistent and severe
Painless jaundice, pruritis, steatorrhorea
DM
Trousseau’s syndrome
Ascites
Abdominal mass
Vrichow’s node
Corvosier’s sign
Troussau’s syndrome
The Trousseau sign of malignancy or Trousseau’s Syndrome is a medical sign involving episodes of vessel inflammation due to blood clot (thrombophlebitis) which are recurrent or appearing in different locations over time (thrombophlebitis migrans or migratory thrombophlebitis). The location of the clot is tender and the clot can be felt as a nodule under the skin.[1] Trousseau’s syndrome is a rare variant of venous thromboembolism (VTE) that is characterized by recurrent, migratory thrombosis in superficial veins and in uncommon sites, such as the chest wall and arms. This syndrome is particularly associated with pancreatic and lung cancer. [2] Trousseau’s Syndrome can be an early sign of gastric or pancreatic cancer,[3] typically appearing months to years before the tumor would be otherwise detected.[4] Heparin therapy is recommended to prevent future clots.[5] The Trousseau sign of malignancy should not be confused with the Trousseau sign of latent tetany caused by hypocalcemia.
Ix in ?pancreatic ductal cell carcinoma?
Bloods: decreased Hb, raised bilirbuin, hypercalcaemia
CT/MRI/ERCP
CA19.9 >70IU
Tumour marker in pancreatic acrinoma?
CA19.9
Mx of pancreatic ductal carcinom
CTx palliative (5FU)
Whipple’s procedure
Px v poor <5% 5YSR
What type of pancreatic malignancy is this?
Ductal cell carcinoma

What are the features of pancreatic endocrine tumours?
Group of epithelial tumours of the pancreas showing endocrine differentiation
May be functioning or non-functioning
2% of all pancreatic tumours
30-60y/o
Associated with MEN1
Symptoms of functional neuroendocrine tumours of the pancreas
Present with symptoms related to hormone excess e.g.
Insulinoma: hypoglycaemic attacks
Gastrinoma: Zollinger-Ellison syndrome: recurrent ulceration
VIPoma: diarrhoea
Glucagonoma: necrolytic migrating erythemia
Ix in neuroendocrine tumour of pancreas
Mx
CT/MRI
Sx
MEN1
PPP
Parathyroid hyperplasia/adenoma
Pancreatic endocrine tumour
Pituitary adenoma
MEN 2A
Parathyroi
Thyroid
Phaeo
MEN2B
Medullary thyroid
Phaeo
Neuroma
Marfanoid phenotype
What are the different types of pancreatic cystic tumours?
Intraductal papillary mucinous neoplasm
Mucinous cystic neoplasm
Serous cystic neoplasms
Solid pseudopapillary neoplasm
What type of pancreatic malignancy is this?

Pancreatic neuroendocrine tumour
What is atherosclerosis?
A stereotypic arterial response to injury caused by an excessive and harmful inflammatory and fibroproliferative response
Chronic inflammation in the intima of large arteries
What are the sites of predilection for atherosclerosis?
Branch points of arteries, bifurcations and curvatures
What is shear stress?
Sideways force on the endothelium representing the tendency to be dragged along with blood flow.
Turbulence is atherogenic
Pathophysiology of atherosclerosis?
Damage to smooth endothelium
LDL enters hte endothelium, becomes trapped and undergoes oxidative modification
Monocyte recruitment via chemokines (MCP-1)
Adhere to endothelium via ICAM-1 and VCAM-1
Transendothelial migration
Oxidised lipoproteins are taken up by monocytes, recognised by macrophage scavenger receptors and PRRs (can stain for macrophages with CD68)
These form foam cells which excrete inflammatory mediators e.g. IL-1
Collagenolysis via MMP occurs
Foam cells die by apoptosis releasing cholesterol into a lipid core.
Fatty streaks are hte earliest lesions seen in atherosclerosis
Platelets stick to the damaged tissue
Endothelium proliferates: fibrous cap forms on top of the endothelium and more cholesterol is deposited in the core
The plaque enlarges blocking arteries
What is the earliest lesion seen in atherosclerosis
Fatty streaks
What is the function of LDL
Cholesterol rich, carries cholesterol from liver to tissues
Oxidised LDL causes inflammation, endothelial damage and VSMC death
What are the functions of VSMCs?
Two phenotypes: contractile and synthetic:
Synthesise collagen and strengthen the fibrous cap
Function of endothelial cells
Biologically active lining of BVs
Portal of LDL emigration
Modualted by shear stress
Express adhesion molecules
Features of familial hypercholesterolaemia?
AR
Extreme cholesterol levels >20mmol/L which is resistant to lipid lowering agents
Xanthomas in skin and tendons
Rapid atherosclerosis
Timescale for atherosclerosis
Intermediate lesions: 20y
Advanced lesions: 40y
Cx >40y
What are the Cxs of atherosclerosis?
Stenosis: carotids
Rupture: leading to sudden thrombosis and or emoblism
Erosion: can lead to thrombosis
Intraplaque haemorrhage
Aneurysm
What are the structures of unstable plaques
Large lipid core, fibrous cap with a prominent inflammatory infiltrate
Inflammation has a lytic effect and there are fewer synthetic VSMCs
There is a structural breakdown of the superficial cap which tears causing intraplaque haemorrhage and superficial endothelial erosion
Symptoms of atherosclerosis
Insufficient blood supply leads to cellular dysfunction which is often experienced as pain on exertion: angina pectoris, intermitten claudication
Critical iscahemia results in cell death: MI, cerebral infarction, gangrene
Cell death occurs, then micrsscopic changes, then gross cahnges
What is usually the cause of death in MI?
VF
MI Histology: <6h
Normal
MI Histology: 6-24h
Loss of nuclei, homogenous cytoplasm and necrotic cell death
Neutrophil infiltration occurs first
MI Histology: 1-4d
Infiltration of polymorphs
Then macrophages- clear debris
MI Histology: 5-10d
Removal of debris
MI Histology: 1-2w
Granulation tissue, new blood vessels, myofibroblasts and collagen synthesis
Weeks-months-> strengthening and decellularising scar
What are the complications of acute MI
Arrythmias
Congestive heart failure
Fibrionous pericarditis
Cardiac rupture
Papillary muscle dysfunction or rupture
Aneurysm
PE
Dressler syndrome
Cardiogenic schock
M
What are the mechanical complications of MI?
Rupture of the LV, rupture of the interventricular septum and MR
What are the pericardial complications of MI
3 types:
Early infarct associated pericarditis
Pericardial effusion (+/- tamponade)
Dressler’s
Why does MI lead to increased risk of arrythmia?
The damaged myocardium acts as a substrate for re-entrant circuits due to changes in tissue refractoriness:
Generalised autonomic dysfunction resulting in enhanced automaticity of the myocardium and conduction system
There is an electrolyte imbalance: hypokalaemia and hypomagnesia
Transmural infarction can interrupt afferent and efferent limbs of the SNS
How does the risk of VF change after MI
Greatest immediately after, declines afterwards
What proportion of patients with AMI are susceptible to arrythmia?
90%
Which type of MI has an increased risk of VF?
STEMI
What is the consequence of papillary muscle dysfunction or rupture
Can cause mitral valve incompetence
Usually occurs in the first few days
When does thromboembolism follow MI?
>1w due to mural thrombi overlying infarcted tissue
How do aneurysms arise in AMI
End-stage scars can form large transmural infarcts which result in large bulging aneurysms
>4w
What is the triad in Dresslers?
Triad of fever, pleuritic pain and pericardial effusion
What are the causes of HF?
IHD
Valve disease
Myocarditis
HTN
Cardiomyopathy
Pathology seen in HF?
Dilated heart
Scarring and thinning of the walls
Pleural effusion
Pulmonary oedema
Hepatomegaly
Ascites
Peripheral oedema
Why does ventricular remodelling occur?
Neurohumoral imbalance, increased cytokine expression, immune and inflammatory changes and altered fibrinolysis. Leads to multiple effects
Hypertrophic heart leads to?
Diastolic heart failure
Dilated heart leads to?
Systolic heart failure
Features of liver in HF
nutmeg liver
Venous congestion and fatty changes
Hepatic cirrhosis with firosis
Def: cardiomyopathy
Intrinsic disease of heart muscle
Def: dilated cardiomyopathy
Progressive loss of myocytes, characterised by a dilated heart, HF, arrythmias and sudden death
Pathology of dilated cardiomyopathy
Myocytes replaced with fine interstitial fibrosis
Causes of dilated cardiomyopathy
Idiopathic
Infective
Toxic: ETOH, CTx, cobalt Fe
Hormonal: hyper, hypothyroid, DM, peri-partum
Genetics: haemochromatosis, Fabry’s, Mc ARdle’s
Immunological: myocarditis
Def: hypertrophic cardiomyopathy
LV hypertrophy, characteristic fibre dissaray
Familial in 50% (AD with variable penetrance)
Thickening of the septum narrows the LV outflow tract
Clinical findings in hypertrophic cardiomyopathy
Systolic murmur
Arrhythmias
Hx of sudden death
Cause of HOCM
Sarcomere mutations (myosin heavy chain)
Def: restrictive cardiomyopathy
Walls are rigid, heart is prevented from stretching and filling with blood
Causes of restrictive cardiomyopathy
Priamry:
Loffler’s endocarditis
Endocardial fibroelastosis
Secondary:
Infiltrative: amyloidosis, sarcoidosis, haemochromatosis
Interstitial: postradiation fibrosis
Loeffler endocarditis
Loeffler endocarditis is a form of restrictive cardiomyopathy which affects the endocardium and occurs with white blood cell proliferation, specifically of eosinophils.[1] Restrictive cardiomyopathy is defined as a disease of the heart muscle which results in impaired filling of the heart ventricles during diastole.[1][2]
Features of chronic rheumatic valve disease?
Sequaelae of earlier rheumatic fever, predominantly left sided
Mitral>aortic>tricuspid>pulmonic
Which valve is most commonly affected in rheumatic fever?
Mitral
What are the pathological chagnes occuring to the valve in chronic rheumatic valvular disease?
Leaflets thicken
Fusion of the commissures
Chordae tendinae also thicken, shorten and fuse
What are Aschoff bodies?
Nodules found in hearts of individuals with rheumatic fever which resulted from inflammation of the heart muscle
What are the causes of AR?
Rigid: rheumatic and degenerative
Destructive: microbial endocarditis
Collapse: Prolapse through a VSD or due to myxomatous degeneration
Disease of the arotic valve ring
Mecahnical: if the heart dilates the valve may become insufficient to cover the dilated area, can occur in Marfan’s dissecting aneurysms, ank spond, syphillitic aortitis and cystic medial degeneration of the aortic media
Presentation of MR
Middle aged woman with SOC, chest pain, mid systolic click, late systolic murmur
Histology of MR
Increased glycosaminoglycans which are prominent on Alvian blue stain
Types of pericarditis?
Fibrinous: MI and uraemia
Purulent: staphylococcus
Granulomatous: TB
Haemorrhagic: tumour, TB, uraemia
Fibours aka constrictive
What is usually the cause of pericardial effusion?
Chronic heart failure
Features of LV failure
Damming of blood within pulmonary circulation_> dyspnoea, orthopnoea, PND, wheeze, fatigue
Eventually leads to reduced peripheral BP and flow
Ix in HF
BNP, CXR, ECG, Echo
What is the most common mutation in HOCM?
Beta MHC
Features of acute rheumatic fever?
Heart: pancraditis
Joints: arthritis and synovitis
Skin: erythema marginatum, subcut nodules
Neurological: Encephalopathy, Sydenham’s chorea
Clinical presentaiton of rheumatic fever
Clinical features develop 2-4w post strep throat
What is the main group causing acute rheumatic fever?
Lancefield GAS
Anitschkov myocytes?
Regenerating myocytes seen in acute rheumatic fever
Verrucae
Aschoff bodies
Anitschkov myocytes
Rheumatic fever
Treatment of acute rheumatic fever
Benpen
Erythromycin if penallergic
What are the causes of vegetative endocarditis?
Rheumatic heart disease
Infective endocarditis
Non-bacterial thrombotic endocarditis (marantic)
Libman Sacks endocarditis
Large irregular masses on valve cusps extending into the chordea
Infective endocarditis
Small, bland vegetations attached to lines of closure, formed of thrombi
Marantic
<2mm vegetations that are sterile and platelet rich
Libman=Sacks Endocarditis
With what is Libman Sacks endocarditis associated?
SLE and anti-phsopholipid
What are the predisposing factors leading to infective endocarditis?
Rheumatic heart disease
Mitral valve prolapse
Calcified valves
Bicuspid aortic valve
Prosthetic valve
Congential defects
What is infective endocarditis usually secondary to?
Bacteraemia as a consequence of
poor dentalo hygiene
IVDU
Soft tissue infection
Dental treatments
Cannulae
Cardiac Sx/pacemeakers
What are the casuative organisms in acute infective endocarditis?
Staph aureus
Strep pyogenes
Pathology of acute infective endocarditis
Larger and more localised vegetations spreading into the aorta
What are the causative organisms in subacute infective endocarditis?
Strep viridans
Staph epidermis
HACEK
Coxiella
Mycoplasma
Candida
Pathology in subacute infective endocarditis?
Friable, soft thrombi, small
Extending into chordae
Clinical features of infective endocarditis
Fever, malaise, anaemia, rigors, splenomegaly, new murmur
Roth spots
Splinter haemorrhages
Janeway lesions
Osler’s nodes
Usually mitral/aortic unless IVDU
Dx of infective endocarditis
Duke’s criteria
Rx in infective endocarditis?
Benpen + gentamicin
Site, pathology, aetiology:
Chronic bronchitis
Bronchus
Dilatation nof the airways and excess mucus production
Tobacco smoke, air pollution
Site, pathology, aetiology:
Bronchiectasis
Bronchus
Airway dilatation and scarring
Site, pathology, aetiology:
Asthma
Bronchus
SM cell hyperplasia, excess mucus, inflammation
Immunologic
Site, pathology, aetiology:
Emphysema
Acinus
Airspace enlargement, wall destruction
Tobacco smoke, alpha-1 anti-trypsin deficiency
Site, pathology, aetiology:
Bronchiolitis
Bronchiole
Inflammatory scarring, obliteration
Tobacco smoke, air pollutants
Clinical features of chronic bronchitis
Cough and sputum on most days for 3m over 2 years
Dilatation of the airways
Goblet cell hyperplasia
Hypertrophy of mucuous glands
Chronic bonchitis
Cx of chronic bronchitis
Reuccrent infections
Chronic hypoxia
Pulmonary HTN
Permanent dilatation of the bronchi
Bronchiectasis
Curschmann spirals
Charcot-Leyden crystals
Asthma
Whorls of shed epithelium
Curschmann spirals
Whorls of shed epithelium
Eosinophils
Charcot-Leyden crystals
Asthma
Loss of the alveolar parnehcyma distal to the terminal bronchiole
Emphysema
Cx of emphysema
Pneumothorax
Respiratory failure
Pulmonary HTN
Cx of bronchiectasis
Recurrent infections
Haemoptysis
Pulmonary HTN
Amyloidosis
Symptoms of bronchiectasis
Cough
Purulent sputum
Fever
Causes of bronchiectasis
Inflammatory:
Post-inefctious
Abnromal host defence: hypogammaglobinaemia, CTx
Ciliary dyskinesia: Kartageners and 2o
Obstruction
Post-inflammatory (aspiration)
Secondary to bronchiolar disease and interstitial fibrosis
Systemic disease
Asthma
Congenital:
CF
Primary ciliary dyskinesia
Hypogammaglobulinaemia
Def: interstitial lung disease
Group of >200 disease characterised by inflammation and fibrosis of the pulmonary connective tissue, particulary the most peripheral and delivate interstitium of the alveolar wall
What are the features of interstital lung disease
Restrictive features on spirometry:
Decreased CO diffusion capacity
Decreased lung volume
Decreased compliance
Presentation of interstitial lung disease
SOB
End-inspiratory crackles
Cyanosis, pulmonary HTN and cor pulmonale
All have honey-comb lung in end stage
Honey-comb lung
Interstitial lung disease
How can interstitial lung disease be characterised?
Fibrosing:
Idiopathic pulmoanry fibrosis
Pneumoconiosis
Crpyotgenic organising pneumonia
Associated with CTD
Drug-induced
Radiation pneumonitis
Granulomatous:
Sarcoid
EAA
Associated with vasculitides e.g. Wegener’s, Churg-Staruss, microscopic polyangitis
Eosinophilic
Smoking related
Features of cryptogenic fibrosin alveolitis/idiopathic pulmonary fibrosis
M>F
Unknown causative agents
Usual intersitital pneumonia required for Dx
(also seen in CTD, asbestosis and EAA)
Progressive patchy, interstitial fibrosis with loss of normal lung architecture and honeycomb change, beginning at the periphery of the lobule, usualy sub-pleural
Hyperplasia of type 2 pneumocytes causing cyst formation= honeycomb
Usual Interstitial fibrosis
Clinical presentation of Idiopathic interstitial fibrosis
Increasing exertional dyspnoea and non-productive cough
40-70y/o
Hypoxaemia-> cyanosis and pulmonary HTN
+/- cor pulmonale and clubbing
Rx of interstitial fibrosis
Steroids
Cyclophosphamide
Azathioprine
(Limited impact on survival)
Which CTDs can have prominent lung fibrosis?
RA, systemic sclerosis, SLE
Def: pneumoconiosis
Typically and occupational lung disease: non-neoplastic reaction to inhalation of mineral dusts or inorganic particles
Majority affect the upper lobe e.g. coal workers penumoconiosis, silicosis, asbsestosis (tends to affect the lower lobe)
Granulomatous infections
TB, fungal (histoplasma, cryptococcus, coccidioides, aspergillus, pneumoystitsi) parasites
Non-infectious granulomatous disease
Sarcoid
Foreign body
Drugs or occupational lung disease
What are the group of immune-mediated lung disorders characterised by intense/prolonged exposure to inhaled organic antigens leading to widespread alveolar inflammation
EEA
Hypersensitivity pneumonitiis
Crypotgenic organisng pneumonia
Bronchiolitis obliterans organising pneumonia
Polypoid plugs of loose connective tissue within alevoli/bronchioes?
Feature of granuloma formation and organisn pneumonia
Acute presentation of EEA etc
Inhalation of antigenic dust in sensitised individual leading to systemic symptoms (fever, chills, chest pain, SOB, cough) within hours of exposure
Usually settles by following day
Progresses to EEA
Progressive, persistent productive cough and SOB
Finger clubbing and severe weight loss
EEA
Different types of EEA
Farmers lung: moudly hay
- Saccharopolyspora rectivirgula
Pigeon fanciers lung
- Proteins in excreta/feathers
Humidifiers lung
- heated reservoirs
Malt-workers lung:
- germinating barley
Cheese washers loung:
- fungi
What are the different categhroies of penumonia?
Bronchopneumonia: patchy bronchial/peri-bronchial distribution. Low virulence organisms
Lobar pneumonis: fibrinosuppurative consolidation
Atypical: interstitial pneumonitiis without intra-alveolar inflammation
Stages of lobar pneumonia
Consolidation
Red hepatisation (neutrophilia)
Grey hepatisation (fibrosis)
Resolution
What are the diseases oif the pulmonary vasculature
PE
Pulmonary HTN
What are the tumours of the lung
Squamous CC
Adenocarcinoma
Small CC
Large CC
Mesothelioma
What is the most common tumour of the lung?
Squamous CC
Which lung cancers are more common in smokers?
Squamous cell
Small cell
Features of squamous cell carcinoma
Closely correlated with smoking
M>F
Usually proximal bronchi, local spread with late metastasis
Less responsive to chemo
Variety of subtypyes: papillary, basaloid,
Associated with caviation and hypercalcaemia
Progression of squamous cell carcinoma
Epithelium
Hyperplasia
Squamous metaplasia
Angiosquamous dysplasia
Carcinoma in situ
Invasive carcinoma
Keratinisation, intercellular prickles
Sqamous cells on cytology
Squamous cell lung carcinoma
Features of lung adenocarcinoma
Most common in womena dn non smokers
Malignant epithelial tumour with glandular differntiation or mucin production
Occurs peripherally and metastasises early
Histo: glandular differentiation
Cyto: cells containing mucin vaculose
EGFR mutations
Adenocarcinoma
Progression of adenocarcinoma
Atytpical adenomatous hyperplasia-> non mucinous BAC-> mixed pattern adenocarcinoma
Features of small cell carcinoma
Usually occurs centrally in the proximal bronchi
Arises from neuroendocrine cellls
Associated with ectopic ACTH secretion, Lambert-Eaton, cerebellar degeneration
Highly malignant with early metastasis usually by diagnosis
Poor prognosis
p53 and RB1 mutations common
Lambert-Eaton
Lambert-Eaton myasthenic syndrome (LEMS) is an autoimmune disease — a disease in which the immune system attacks the body’s own tissues. The attack occurs at the connection between nerve and muscle (the neuromuscular junction) and interferes with the ability of nerve cells to send signals to muscle cells.
Paraneoplastic cerebellar degeneration syndrome
Paraneoplastic syndromes are a group of rare disorders that are triggered by an abnormal immune system response to an underlying (usually undetected) malignant tumor. Patients with paraneoplastic neurological syndrome (PNS) most often present with neurologic symptoms before an underlying tumor is detected.
Paraneoplastic neurologic syndromes include many neurologic disorders including paraneoplastic cerebellar degeneration (PCD) caused by an immune-mediated mechanism other than a metastatic complication in patients with an underlying malignancy. Any part of the nervous system can be involved depending on the type of primary malignancy. These syndromes affect 1-3% of all cancer patients.[1] These syndromes are difficult to diagnose and respond poorly to treatment. However, the oncologic outcome of patients with antibody-associated paraneoplastic syndromes does not significantly differ from that of patients who do not have the antibodies or a paraneoplastic syndrome.
Anti-Yo
Paraneoplastic cerebellar degeneration
Features of large cell carcinoma
Poorly differentiated malignant epithelial tumour: large cells, large nuclei with prominent nuceloli
No evidence of glandular or squamous differentiation
Associated with paraneoplastic syndromes:
ADH, ACTH, PTH, PTHrP, Calcitonin, serotonin, bradykinin
NSCLC: ERCC1
Poorer response to cisplatin
EGFR in adenocarcinoma
Target for anti-EGFR TKI therapy
Kras mut in adeno/squamous cell carcinoma
Poor prognosis- no response to TKI
EML4-ALK
Usualy adenocarcinoma= no benefit from TKI
Staging of lung malignancy
T1-4 based on size, invasion of pleura and pericardium
LN metas: N0-2 (N0- no LNs, N1-N2 LNs involved, dependant on extent)
M0-1: mets or na
Features of mesothelioma
Arises from either parietal or visceral pleura
Spreads within pleural space
Associated with pleural effusion, chest pain and dyspnoea
Latent period of 25-45 following asbestos exposure
How do large PEs affect pulmonary vasculature
Impact in the main pulmonary arteries leading to acute cor pulmonale, cardiogenic shock, and death if >60% of pulmonary bed occluded
Saddle embolus=
Occluding the pulmonary trunk
How do small emboli impact pulmonary vasculature
May be silent or cause peripheral wedge infarctions
Repeated infarctions can lead to pulmonary HTN
What are non-thrombotic emboli
BM, amniotic fluid, tumour, air, foreign body
Def: pulmonary HTN
Mean pulmonary arterial pressure >25mmHg at rest
Causes of pulmonary HTN
Can be primary
More often secondary due to:
Chronic lung disease
Heart diseases
Recurrent thrombo-emboli
Autoimmune disorders
What are the group most commonly affected by primary pulmonary HTN?
Women aged 20-40
How can secondary causes of pulmonary HTN be classified?
Pre-capillary:
Chronic hypoxia/embolus
Capillary:
Fibrosis
Post-capillary:
Left heart disease, veno-occlusive disease
Def: pulmonary oedema
Intra-alvolar fluid accumulation leading to poor gas exchange, main aetiology is left heart failure
Iron-laden macrophages=
Heart failure cells
Seen in pulmonary oedema due to heart failure
Causes of diffuse alveolar damage
ARDS in adults
Neonates: hyaline membrane disease of newborn, insufficient surfactant production
Common congenital disorders of the lung?
Lung agenesis or hypoplasia
Tracheal and bronchial stenosis
Congeital cysts
Causes of ARDS
Infection
Aspiration
Trauma
Inhaled irritant gases
Shock
Blood transfusion
DIC
Drug overdose
Pancreatitis
Idiopathic


Macroscopic features of asthma?
Mucus plug
Overinflated lungs
What are the two types of emphysema and their causes?
Paracinar: caused by anti-trypsin deficiency
Centrilobular: caused by smoking, loss of the centre of the bronchiole
What are the organs affected by CF?
Lung
GIT: meconium ileus and malabsroption
Pancreas: pancreatitis and malabsorption
Liver: cirrhosis
Male reproductive system: infertility
How does chronic hypoxia cause pulmonary HTN
Normal response of lung is to reduce blood flow to hypoxic areas
Results in chronic vasoconstriction of the pulmonary arterioles which leads to morphological changes e.g. eccentric intimal fibrosis, thickening of the muscle wall
What is pulmonary veno-occulsive disease
Fibrotic occlusion of the pulmonary veins can be caused by:
Idiopathic
Herbal teas and diet pills
CTx
RTx
BM transplant
Renal transplant
HIV
Systemic sclerosis
Dx of idiopathic pulmonary fibrosis
HRCT +- biopsy
What is an issue with bevacizumab in squamous cell carcinoma
Some patients develop fatal haemorrhages with some durgs as it is an angiogenesis inhibitor
Stain for adenocarcinoma
TTF-1
Stain for SqCC
CK5/6, P63, TTF-1
Cx of lung cancer
Bronchial obstruction
Invasion of local structures
Extension through pleura or pericardium
Diffuse lymphatic spread within lungs
Metabolic spread
Paraneoplastic ssyndromes
What are the paranetoplastic syndromes seen in lung cancer and their consequences?
ADH: SIADH causing hyponatraemia
ACTH: Cushings
PTHrP: hypercalcaemia
Calcitonin; hypocalcaemia
Gonadotrophin: gynaecomastia
Serotonin: carcinoid syndrome
Nonednocrine: haematological/coagulation defects
Consequences of lung cancer invading local structures
SVCO
Oesophagus: dysphagia
Horner’s syndrome
Pathology in chronic meningitis?
Leptomeninges and sometimes dura thickened
Arachnoid adhesions may be present
Lymphocyte, plasma cell and epitheloid macrophages seen in the infiltrate
Caseous necrosis and granulomatous inflammation may be present in TB
Adhesions can cause a non-communicating hydrocephalus
What are the leptomeninges?
Pia and arachnoid mater and the subarachnoid space
Inflammation usually due to infection.
Features of cystitis
Can be acute or chronic
85% caused by gram negative bacilli that are normally resident in GIT e.g. E Coli etc
What drug is associated with cystitis?
Cyclophosphamide with haemorrhagic cystitis
What are the risk factors for cystitis
Female
Calculi
Urinary obstruction
DM
Sexually active
Instrumentation
(Cyclophosphamide)
What are the types of bladder tumours?
Transitional cell
Squamous cell
Adenocarcinoma
What is the most common bladder malignancy?
Transitional cell
Epidemiology of TCC
90% of all bladder tumours
Male>Female
50-80y/o
Risk factors for bladder TCC
Cigarette smoking
Aromatic amines
Clinical features of TCC
Painless haematuria, freuqency, urgency, pyelonephritis or hydronephrosis if ureteral orifices involved
Dx of TCC
Cystoscopy + biopsy
With what is squamous CC of the bladder associated?
Schistosomiasis
What is the aetiology of bladder adenocarcinoma?
Rare, arising from extensive intestinal metaplasia or from urachal remnants
What is the urachus?
The urachus is a remnant of a channel between the bladder and the umbilicus (belly button) where urine initially drains in the fetus during the 1st trimester of pregnancy. The channel of the urachus usually seals off and obliterates around the 12th week of gestation and all that is left is a small fibrous cord between the bladder and umbilicus called the median umbilical ligament.
What is BPH
DHT-mediated hyperplasia of prostatic stromal and epithelial cells resulting in the formation of large nodules
Symptoms of BPH
Difficulty urinating
Retention
Frequency
Nocturia
Overflow dribbling
Features of chronic pyelonephritis and reflux nephropathy
Tubulo-interstitial inflammation causing scars, leads to blunting of the renal calcyes and deformation,
Xanthogranulomatous PN can result from Gram -ve infections
Mx of BPH
TURP
5 alpha reductase inhibitors
What is the most common form of prostate Ca
Adenocarcinoma
Commonly seen in older gentlemen
What is the scoring system for prostate carcinoma?
Gleason scoring:
Pattern 1- well differentiated tumours, uniform round glands packed into circumscribed nodules
Pattern 5: no glandular differentiation
Final score= predominant pattern plus worst pattern
Gleason grading
Grade 1 <6
Grade 2 6+7
Grade 3 >8
Dx of prostate adenocarcinoma
Hx
Ex
PSA (.4ng/ml)
Mx of prostate adenocarcinoma
Stage T1/T2: RTx and Sx
T3: Extraprostatic spread: external beam radiation
Causes of acute prostatitis?
E Coli and other gram -ve rods
Fever, chills and dysuria
Features of chronic abacterial prostatitis
Manifestation similar to chronic bacterial but without hx of recurrent UTI
Culture negative
Dysuria or mild suprapubic discomfort
Chronic bacterial prostatitis?
Symptoms of recurrent UTI
What is the most common cause of granulomatous prostatitis?
Bladder BCG for treatment of carcinoma in situ
Non-specific granulomatous prostatitis
Symptoms of urinary incontinence and pelvic discomfort
What are the most common types of testicular tumours?
Germ cells
Draw the types of testicular tumours

What is the most common type of germinal tumour?
Seminoma
What is the impact of undescended testes on cancer risk?
10x
What is PIN
Prostatic intraepithelial neoplasia
Common in young men
33% risk of carcinoma
Doesn’t cause an elevated PSA but examine for carcinoma
What are the markers for germ cell tumours?
AFp
beta HCG
LDH
Seminoma: age group
15-34y
Seminoma marker?
Beta HCG
(AFP and ALP not raised)
What is the most common testicular carcinoma in <3y/o?
Embryonal carcinoma and yolk sac tumour
Aggressive and malignant
AFP
Marker of endodermal sinus tumour?
AFP
Marker of choriocarcinoma?
HCG
What is a condyloma acuminatum?
Genital wart
What is the malignant penile lesion?
Squamous cell carcinoma
HPV-related
Clinical features of renal cell carcinoma?
Costovertebral pain
Palpable mass
Haematuria
Paraneoplastic syndromes: Polycythaemia, hypercalcaemia, HTN, Cushing’s, amyloidosis
Risk factors for testicular tumours?
Cryptorchidism
Testicular dysgenesis
Klinfelters
Testicular feminisation
What are the types of renal carcioma
Clear cell
Papillary: commonest in dialysis-associated cystic disease
Chromophobe renal carcinoma: pale eosinophilic cells
What are the most common organisms causing PID
Chlamydia
Gonorrhoea
Symptoms of PID
Lower abdo pain
Dyspareunia
Vaginal bleeding/discharge
Fever
Adnexal tenderness
Cervical excitation
Fitz-Hugh Curtis
RUQ from perihepatitis
Violin string peri-hepatic adhesions
Cxs of PID
FHC syndrome
Infertility
Increased risk of ectopic
Intestinal obstruciton-> bacteraemia
Tubo-ovarian abscess
Peritonitis
Plical fusion
Def: endometriosis
Presence of endometrial glands or stroma outside of uterus
10% of women
What are the 3 theories around the aetiology of endometriosis?
Metaplasia of pelvic peritnoum
Implanatation of endometrium through retrograde menstruation
Induction of metaplasia
Pelvic pain
Dysmenorrhoea
Deep dyspareunia
Reduced fertility
Nodules/tenderness in posterior fornix or uterus
Immobile uterus which is anteverted in severe disease
Endometriosis
Red-blue to brown nodules “powder burns”
Choclate cysts in ovaries
Endometriosis
Endometriomas
Def: adenomysosis
Ectopic endometrial tissue deep within the myometrium causing dysmenorrhoea
May cause menorrhagia and deep dyspareunia
Globular uterus
What is the most common tumour of the female genital tract?
Leiomyoma
Draw the location of fibroids

What is important re fibroids
Oestrogen stimulation:
Enlarge during pregnancy
Regress post-menopausally
Macroscopic and microscopic features of leiomyomas?
Sharp, circumscribed, discrete, round, firm, grey-white tumours of variable size
Bundles of smooth muscle cells
Clinical features of fibroids
Menorrhagia
Dysmenorrhoea
Pressure effects: urinary frequency, tenesmus
Subfertility
Red degeneration in pregnancy
May rarely transform to leiomyosarcoma
Leiomyosarcoma aetiology
May be due to transformation of leiomyoma
More common post-menopausally
How is endometrial carcinoma staged?
FIGO
Stage 1: confined to uterus
Stage 2: involves cervix
Stage 3: spread to adenezae, serosa, positive peritoneal cytlogy, LNs (pelvic or para-aortic)
Stage 4: pelvic organs and distal spread
Draw the division of endometrial cancer types

What are the features of endometroid carcinoma
Look similar to normal endometrial glands
Associated with oestrogen excess, uusally in peri-menopausal women
Risk factors for endometroid endometrial carcinoma
E2 excess:
OBESITY
Anovulatory amenorrhoea (PCOS)
Nulliparity
Early menarche
Late menopause
Tamoxifen
DM
HTN
What is the more aggressive type of endometrial carcinoma?
Non-endometroid
Features of VIN
Dysplasia of epithelium associated with HPV
I, II and III
Progression to invasive disease is lower than for CIN
Significanece of Paget’s disease of vulva?
Adenocarcinoma in situ, rarely associated with invasive adenocarcinoma
Tissue type in VIN
Generally squamous cell
Draw the organisaiton of ovarian tumours

Psammoma bodies, columnar epithelium=
Serous epithelial tumour
No psammoma bodies
Associated with pseudomyxoma peritonei
Mucinous tumour of ovary
Clear cytoplasm with hobnail appearance
Clear cell epithelial tumour of ovary
What is the female counterpary of the seminoma
Dysgerminoma
What is a mature teratome?
Dermoid cyst
Contains skin, hair, teeth, etc.
What are immature teratomas
Germ cell tumours of the ovary
Malignant and usually solid, contain immature, embryonal tissues
Meig’s syndrome associated with?
Fibroma of ovary
Which ovarian malignancy produces E2?
Granulosa-Theca cell tumour
Which ovarian malignancy produces androgens?
Sertoli-Leydig tumour
What are the secondary ovarian tumours?
Endometroid, serousus, clear cell carcinomas: may be uterine primary
Krukenbergy tumours: metastatic mucin producing adenocarcinomas from stomach or breast
Eponymous name for metastatic mucin-producing adenocarcinoma from stomach/breast
Krukenberg tumour
What HPV strains associated with CIN
Commonly 16 and 18
What is the transitional zone
Where the squamous cell transforms into columnar epithelium
CIN I-III
I: dysplasia confined to lower 1/3
II: lower 2/3
III: full thickness but with bm intact
Px of CIN grades
I: reverts to normal
30% of 3 progresses to cervical cancer over 10 yeras
What is CGIN
Less common, treatment requires excision of entire endocervix
What is the most common form of cervical carcinoma
Squamous cell although 20% can be adenocarcinomas etc.
What marks the change from CIN to carcinoma
Invasion through basement membrane
Clinical features of cervical carcinoma
PCB
IMB
Post MB
Pain
Features of SLE
Autoimmune multi-system disorder
Type III hypersensitivity reaction
Increased in classical complement deficiciencies.
Can be drug-induced
Higher prevalence in Afrocarribean and women
What HLA is associated with SLE
HLA DR3 (or 2)
Autoantibodies in Lupus
ANA (95%)
Anti dsDNA
Anti-Sm
Drug-induced
Anti-histone
LE bodies
Small vessel angiopathy
Onion skin lesions in the spleen
Libman-Sack endocarditis
Lupus
Signs and symptoms of Lupus
SOAP
BRAIN
MD
Serositis
Oral ulcers
Arthritis
Photosensitivity
Blood disorders (AIHA, ITP, leucopenia)
Renal involvement
ANA +ve
Immune phenomena (dsDNA, anti-SM, antiphospholipid Ab)
Neuro symptoms
Malar rash
Discoid rash

Lupus
What are the two types of scleroderma?
Limited cutaneous (CREST)
Diffuse scleroderma
Features of scleroderma
Autoimmune multi-system disorders
SKin fibrosis
HLA associations in scleroderma
HLA DR5 and DRw8
Autoantibody in CREST (limited cutaenous)
Anti-centromere
Anti-centromere
CREST (limited cutaenous)
Increased collagen in skin and organs
Onion skin thickening of arterioles
CREST
Signs and symptoms of CREST
Skin changes on face and distal to elbows and knees
Calcinosis
Raynauds
Esophageal dysmotility
Sclerodactyly
Telangiectasia
Associated with pulmonary HTN

Sclerodactyl
CREST
Lung disease in scleroderma
CREST= pulmonary HTN
Diffuse= pulmoanry fibrosis
Autoantibodies in diffuse scleroderma
Anti Scl-70
Fibrillarin
RNA pol I, II, III
PM-SCl
Anti Scl-70
Fibrillarin
RNA pol I, II, III
PM-SCl
Diffuse scleroderma
Inflammation within or around muscle fibres
Diffuse scleroderma
Signs and symptoms of diffuse scleroderma
Skin changes can occur anywhere
Widespread organ involvement
Associated with pulmonary fibrosis
Features of polymyositis/dermatomyositis
Autoimmune inflammatory disorder of muscle +/- skin
Associated with underlying malignancy
Autoantibody in dermatomyositis?
Anti-Jo1
Anti-Mi2
Anti-Jo1
Dermatomyositis
Autoantibody in polymyositis
Anti-signal recognition peptide Ab
(anti-Jo1/ anti-Mi2 (D>P))
Endomysial inflammatory infiltrate
Dermatomyositis
Drop out of acpillaires and myofibre damage
Dermatomyositis
Symtpoms and signs of polymyositis
Proximal limb, anterior neck weakness and oesophageal and respiratoy muscle involvement.
Elevated skeletal muscle enzymes
Abnormal EMG
Positive muscle biopsy
Associated with pulmonary fibrosis
Signs and symptoms of dermatomyositis
Proximal limb, anterior neck weakness and oesophageal and respiratoy muscle involvement.
Elevated skeletal muscle enzymes
Abnormal EMG
Positive muscle biopsy
+ skin changes:
Heliotrope rash
Gottron Papules
Assoc c. pulmonary fibrosis

Gottron papules
Dermatomyositis

Heliotrope rash
Dermatomyositis
What complement defects associated with lupus?
Defects in the classical pathway
What drugs can induce lupus?
Hydralazine
Pocainamide
How is lupus diagnosed
>4 of the ACR criteria
Ix used in Lupus
CRp
ESR
Test for ANA
Complement
Complement in active lupus
C3 (common): Normal
C4 (classical): low i.e. undetectable
Complement in severe active SLE
C3 (common): Low
C4 (classical): low
Complement in inactive SLE
C3: normal
C4: normal
CRP in Lupus
Generally normal
ESR in lupus
High
Anti-dsDNA in Lupus
High
Can be used for disease monitoring
Anti-cardiolipin
Anti-Ro, La, Sm or RNP
in SLE
Sometimes high
Anti-topoisomerase=
Anti-SCl70
What are the large vessel vasculitides?
Takayasu’s
Temporal arteritis
What are the medium vessel vasculitides?
Polyarteritis Nodosa
Kawasaki’s
Buerger’s disease
What are the small vessel vasculitides?
Wegener’s
Churg Strauss
Microscopic polyangitis
HSP
Features of Takayasu’s arteritis
Pulseless disease
Increased in Japanese women
Vascular symptoms: absent pulse, bruits, claudication
Granulomatous vasculitis with massive intimal fibrosis
Pulseless disease
Increased in Japanese women
Vascular symptoms: absent pulse, bruits, claudication
Granulomatous vasculitis with massive intimal fibrosis
Takayasu’s
Features of temporal arteritis
Elderly
Scalp tenderness, temporal headache
Jaw claudication, blurred vision
ESR raised
Overlap with polymyalgia rheumatica
Elderly
Scalp tenderness, temporal headache
Jaw claudication, blurred vision
ESR raised
Overlap with polymyalgia rheumatica
Temporal arteritis
Granulomatous transmural inflammation and giant cells and skip lesions
Temporal arteritis
What is the most common form of arteritis?
Temporal
Features of PAN
Renal involvement is main feature
Can involve other ograns
Spares lungs
30% have underlying Hep B
Renal involvement is main feature
Can involve other ograns
Spares lungs
30% have underlying Hep B
PAN
Microaneurysms on angiography
PAN
Necortising arteritis with inflammation, infiltration of polymorphs, lymphocytes and eosinophils
Arteritis is focal and sharply demarcated
Healed by fibrosis
PAN
Features of Buerger’s disease
Heavy smokers, usually men <35
Inflammation of arteries of extremities: usually tibial and radial
Pain, ulceration of toes, feet, fingers
Corkscrew appearance on angiogram
Heavy smokers, usually men <35
Inflammation of arteries of extremities: usually tibial and radial
Pain, ulceration of toes, feet, fingers
Corkscrew appearance on angiogram
Buerger’s disease
Features of Wegener’s granulomatosis
Upper resp tract: sinusitis, epistaxis, saddle nose
Lower resp tract: cavitation, pulmonary haemorrhage
Kidneys: cresecenteric glomerulonephritis
cANCA +ve
Triad in Wegeners
Upper RT
Lower RT
Kidneys
Saddle nose
Pulmonary haemorrhage
Crescenteric GN
Wegener’s
cANCA +ve
Wegener’s
Churg Strauss
Asthma, allergic rhinitis, eosinophilia
Later systemic involvement
pANCA (anti-PR3) +ve
Asthma, allergic rhinitis, eosinophilia
Later systemic involvement
Churg-Strauss
pANCA (anti-PR3) +ve
Churg Strauss
Microscopic polyarteritis
Granulomatosis with polyangitis
Wegener’s
Eosinophilic granulomatosis with polyangitis
Churg Strauss
What are the 3 types of ANCA-associated vasculitis
Microscopic polyangitis
Wegener’s
Churg Strauss
Features of microscopic polyangitis
Pulmonary renal syndrome:
Pulmonary haemorrhage
Glomerulonephritis
Features of HSP
IgA mediated vasculitis
Affects children
URTI precedes
Palpable purpuric rash, abdo pain, GN, arthritis, Orchitis
IgA mediated vasculitis
Affects children
URTI precedes
Palpable purpuric rash, abdo pain, GN, arthritis, Orchitis
HSP
Features of Cryoglobulinaemia
Presence of abnormal proteins in the blood which becomes thick or gel-like in cold temperatures
Cryoglobulins are Abs
Cause a range of Sx from skin rashes to kidney failure
What is amyloidosis?
Multisystem disorder caused by abnormal protein folding that are subsequently deposited as amyloid fibrils in tissues disrupting their function
How can amyloidosis be classified?
Primary and Secondary
What protein involved in primary amyloidosis?
AL
Derived from light chains therefore more common in patients with myeloma
What protein involved in secondary amyloidosis?
AA
Derived from serum AA which is an acute phase protein
Therefore secondary to chronic infection/inflammation
Associations of primary amyloidosis
Most common form
Most associated with plasma cell dyscrasias
Most have Bence Jones protein in urine and increased plasma cells
Causes of secondary amyloidosis
Autoimmune disease: RA, Ank Spond, IBD
Chronic infections: TB, osteomyelitis, IVDU
Non-imune: RCC, HL
What is seen in haemodialysis associated amyloidosis?
Deposition of beta2-microglobulin
What is the most common form of familial amyloidosis?
Familial Mediterranean Fever
Features of Familal Mediterranean Fever?
++++IL-1 production leading to attacks of fever and inflammation of serosal surfaces
AA amyloid, with predominant renal deposition
Clinical features of amyloidosis?
Caused by amyloid deposits in various organs:
Kidney: nephrotic syndrome
Heart: conduction defects, heart failure
Liver/spleen: hepatosplenomegaly
Tongue: macroglossia
Neuropathies: carpal tunnel
Apple green birefringence with Congo red stain under polarised light?
Amyloidosis
If not polarised light appears pink/red
Def: sarcoidosis
Multisystemic disease of unknown cause
Commonly affects young adults
Characterised by non-caseating granulomas in many tissues
Non-caseating granulomas, Schaumann and asteroid bodies
Sarcoidosis
Features of sarcoidosis
Most severe disease in Afro-Carribeans
Lungs most commonly involved
Often detected at routine CXR
Most seek help with pulmonary symptoms

Bilateral hilar lymphadenopathy
May also see fine nodular shadowing in mid zones
Sarcoidosis
What are the extrapulmonary manifestations of sarcoidosis
Skin: erythema nodosum, lupus pernio, skin nodules
LNs: lymphadenopathy, painless and rubbery
Joints: arthritis, bone cysts
Eyes: anterior uveitis-> misting of vision and painful red eye
Posterior uveitis-> progressive visual loss
Hepatosplenomegaly
Leukopaenia/anaemia
Hypercalcaemia/hpercalcuria
Myocardial involvement
CNS involvement
Keratoconjuncitvits sicca and lacrimal gland enlargement
Symptoms of anterior uveitis
Misting of vision and painful red eye
Symptoms of posterior uveitits
Progressive visual loss
Dx of sarcoidosis?
Dx of exclusion
Raised Ca
Raised ESR
Raised ACE
Transcbronchial biopsy

Lupus pernio
ACE in sarcoid
ngiotensin converting enzyme (ACE) participates in the renin cascade in response to hypovolemia. Its peptidase action on the decapeptide angiotensinogen I results in the hydrolysis of a terminal histidyl leucine dipeptide and the formation of the octapeptide angiotensin II, a potent vasoconstrictor that increases blood pressure.
The primary source of ACE is the endothelium of the lung. ACE activity is increased in sarcoidosis, a systemic granulomatous disease that commonly affects the lungs. In sarcoidosis, ACE is thought to be produced by epithelioid cells and macrophages of the granuloma.
Currently, it appears that ACE activity reflects the severity of sarcoidosis: 68% positivity in those with stage I sarcoidosis, 86% in stage II sarcoidosis, and 91% in stage III sarcoidosis. Serum ACE also appears to reflect the activity of the disease; there is a dramatic decrease in enzyme activity in some patients receiving prednisone.
Other conditions such as Gaucher disease, leprosy, untreated hyperthyroidism, psoriasis, premature infants with respiratory distress syndrome, adults with amyloidosis, and histoplasmosis have been associated with increased levels of ACE.

http://www.lab.anhb.uwa.edu.au/mb140/addons/mcqquiz.htm
Try it
What are the features of acute mastitis
Painful red breast, fever
Almost always occur during lactation and are due to staphylococcal infection via cracks in the nipples
Involved breast tissue is necrotic and infiltratred by neutrophils
Mx of acute mastitis?
Continued expression of milk
Abx
+/- surgical drainage
Features of periductal mastitis
Mostly in smokers and is not associated with lactation
Keritanising squamous epithelium extending deep into nipple duct orificies
Periductal mastitis
Features of duct ectasia
Inflammation and dilatation of the large breast ducts
Poorly defined palpable periarolar mass with thick white nipple secretions
Caused by granulomatous inflammation and dilation of large breast ducts
Mimics mammographic appearance of cancer
Features of fat necrosis
Inflammatory reaction to damaged adipose tissue
Presents as painless breast mass/skin thickening/mammographic lesion
Caused by trauma, surgery, radiotherapy
What are the benign proliferative breast conditions
Fibrocystic disease/ fibroadenosis
Gynaecomastia
What are the changes in breast tissue seen in fibrocystic disease?
Cystic change: small cysts form by dilation of lobules, contain fluid and often calcified
Fibrosis: inflammation and fibrosis secondary to cyst rupture
Adenosis: increased number of acini seen per lobule
Exagerrated response to hormonal influence
Breast lumpiness
Fibrocystic disease
Features of gynaecomastia
Unilateral or bilateral enlargement of the male breast
Indicative of hyperestrinism: ETOH, age, liver cirrhosis, functioning testicular tumour
Epithelial hyperplasia with finger-like projections into ducts
What are the benign proliferative conditions of the breast
Fibroadenoma
Duct papilloma
Radial scar
(Phyllodes tumour)
Features of fibroadenoma
Most common benign tumour, from stroma, often multiple and bilateral
Occuring at any age within the reporudcitve period
Epithelial response to size therefore increase during pregnancy and calcify after menopause
Mobile, well circumscribed, spherical, rubbery breast lump in young woman
Fibroadenoma
Features of duct papilloma
Benign papillary tumour arising within the duct system of the breast
Can be peripheral (within small terminal ductules) or central (larger lactiferous ducts)
Causes bloody discharge, not seen on mammogram, Ix with galactogram
Mx of intraductal papilloma
Excision of the affected duct is curative
Features of Radial scar
Benign sclerosing lesion of the breast
Central scarring surrounded by proliferating glandular tissue in stellate pattern
Resembles cancer on mammogram
Complex sclerosing lesion of the breast
Radiat scar
What are the proliferative breast diseases?
A diverse group of intraductal proliferative lesions of the breast associated with an increased risk of the development of subsequent invasvie breast carcinoma
Usually microscopic asymptomatic lesions
Usual epithelail hyperplasia
Fat epithelial atypia: low grade precursor to DCIS
In situ lobular neoplasia
What are the risk factors for breast carcinoma
Susceptiblity genes
Hormone exposure
Age
FHx
Race: caucasian>Afrocarribean>asian>hispanic
Obesity, tobacco, alcohol
What are the susceptibility genes associated with breast cancer
BRCA1/BRCA2
Increased risk of ovarian, prostate and pancreatic malignancy
BRCA mutations cause a lifetime risk of breast carcinoma of up to 85%
How does hormone exposure impact on breast cancer
Early menarche
Late menopause
Late 1st live birth (pregnancy leads to terminal differentiation of molk-producing luminal cells removing these from the potential cancer pool)
OCP/HRT
Presentation of breast cancer
Hard fixed lump
Paget’s disease of breast
Peau d’orange
Nipple retraction

Paget’s disease of breast
Breast cancer screening programme
47-73 y/o women invited every 3 years for mammography
Features of DCIS
Neoplastic intraductal epithelial proliferation with an inherent but not inevitable risk of progression to breast cancer
Limited to ducts/lobules by basement membranes
Increased incidence since mammography
Appear as areas of microcalficiation
Features of LCIS
Always incidental finding on biopsy as no microcalcifications or stromal reactions
20-40% bilateral
Cells lack adhesion protein
Features of invasive breast carcinoma
Malignant epithelial tumours which infiltrate hte breast with capacity to metastasise
Low grade BCA
Arises from low grade DICS or in situ lobular neoplasia and show 16q loss
What are the different histological classifications of invasive breast cancer
Ductal
Lobular
Tubular
Mucinous
Features of invasive ductal carcinoma
Carcinoma that cannot be classified into another group, most common
Features of invasive lobular carcinoma
Cells aligned in single file chains/strands
Features of tubular carcinomas
Well-formed tubules with low grade nuclei, rarely palpable as <1cm
Features of mucinous breast carcinoma
Cells produce abundant quantities of extracellular mucin which dissects into surrounding stroma
What are the components of the triple assessment?
Examination
Radiological examination (mammography, FNA, USS)
FNA and cytology
What are the components examined in histological grading of breast cancer?
Nuclear pleomorphism, tubule formation, mitotic activity
What happens with all breast neoplasms
Investigated for receptor status:
ER/PR: good prognosis- tamoxifen
Her-2: bad prognosis
What is the single most important factor in px of breast cancer
Axillary LN status
Tamoxifen=
Mixed agonist/antagonist of ER
Trastuzumab
Herceptin
Monoclonal Ab vs Her2
What is an important consideration for Herceptin?
Has a direct toxic effect on myocardium
Must monitor LVEF
Features of basal-like carcinoma of breast
Sheets ot atypical cells with lymphocytic infilrtate
Stains positive for CK5/6/14
Features of Phllodes tumour
Arise from interlobular stroma with increased cellularity and mitosies
>50y with a palpable mass
Low or high grade lesions.
Most benign but can be aggressive
Therefore excised with wide margins to limit local recurrence
Anterior pituitary secretions


What are the symptoms of pituitary disease?
•Hyperpituitarism
–Excess secretion of trophic hormones
–Usually due to functional adenoma
•Hypopituitarism
–Deficiency of trophic hormones
•Local mass effects
What is the most common type of pituitary adenoma?
Prolactinoma
Pituitary microadenoma=
<1cm
What are the clinical effects of prolacitnomas
Amenorrhea, galactorrhoea, loss of libido, infertility
Clinical effects of growth hormone adenomas
Prepubertal children- gigantism
Adults- acromegaly
DM, muscle weakness, HTN, congesitve cardiac failure
Clinical effects of corticotroph cell adenomas
Cushins
What are the significant causes of hypopituitarism
Nonsecretory pituitary adenomas
Ischaemic necrosis:
Sheehan’s
DIC, SCA, elevated ICP, Shock
Pituitary ablation by sx or irradiation
What are the clinical manifestations of anterior pituitary hypofunction
- Children - growth failure (pituitary dwarfism)
- Gonadotrophin deficiency - amenorrhea and infertility in women. Decreased libido and impotence in men
- TSH and ACTH deficiency - hypothyroidism and hypoadrenalism
- Prolactin deficiency - failure of post-partum lactation
What are the peptides released by the posterior pituitary
ADH
Oxytocin
What are the local mass effects of pituitary tumours
Compression of optic chiasm leading to bitemporal hemianopia
Signs and symptoms of raised ICP
Obstructive hydrocephalus

Anterior pituitary

thyroid

Thyroid
Physiology of the thyroid
Responds to TSH from ant pit
Follicular cells convert thyroglobulin into T3 and T3
Effect is to increase BMR
Also contain a population of C -cells (parafollicular) that promote Ca absorption by the skeletal system
Features of non-toxic goitre
Enlargement of the thyroid
Common if there is impaired synthesis of thyroid hormone, most often due to I deficiency
Can be:
Seen at puberty in females
Due to ingestion of substances that interfere with thyroid hormone synthesis
Due to hereditary enzyme defects
Features of multinodular goitre
- With time simple thyroid enlargement may be transformed to a multinodular pattern
- May reach massive size
- May lead to mechanical effects including dysphagia and airways obstruction
- A hyperfunctioning nodule may develop leading to hyperthyroidism
Def: thyrotoxicosis
•Hypermetabolic state caused by elevated circulating levels of free T3 and T4
How can thyrotoxicosis be classified?
Primary:
Grave’s
Hyperfunctioning multinodular goitre
Hyperfunctioning adenoma
Thyroidits
Secondary:
TSH secreting pituitary adenoma (rare)
Not associated with thyroid disease:
Struma ovarii (ovarian teratoma with ectopic thyroid)
Factitious thyrotoxicosis (exogenous thyroid intake)
What is the most common cause of endogenous hyperthyroidism
Grave’s
Triad in Grave’s?
Thyrotoxicosis
Infiltrative opthalmoapthy with exopthalmos
Infiltrative dermopathy (myxoedema)
F>M

Exopthalmos

Pretibial myxoedema
Pathogenesis of Grave’s disease
Autoimmune disorder caused by autoantibodies including those vs TSHR and thyroglobulin
Abs vs TSHR most important-> stimulate release of thyorid hormones
Associated with other autoimmune diseases (hunt in packs)
How can hypothyroidism be classified
Primary:
Postablative
Autoimmune- Hashimoto’s
Iodine deficiency
Congenital biosynthetic defect
Secondary:
Pituitary or hypothalamic failure
What is the most common cause of hypothyroidism
Hashimotos’
Hashimoto’s thyroiditis
What is suggestive of thyroid maligiance in terms of the nature of thyroid nodules
Number- solitary
Consistency- solid
Age of patient- younger
Sex- Male
Iodine uptake- Cold
Features of thyroid adenomas
- Usually solitary
- Well circumscribed lesion that compresses the surrounding parenchyma
- Well formed capsule
- Small proportion cause thyrotoxicosis
- Important to examine the capsule for invasion to exclude follicular carcinoma
Features of papillary carcinoma
Nonfunctional
Painless mass in neck
Cervical mets
10y survival up to 90%
- May occur at any age
- May have papillary architecture
- BUT diagnosis is based on nuclear features
–Optically clear nuclei
–Intranuclear inclusions
•May be psammoma bodies

Papillary carcinoma
Features of follicular carcinoma
- Peak incidence in middle age
- Follicular morphology
- May be well demarcated with minimal invasion or clearly infiltrative
- Usually metastasise via bloodstream to lungs bone and liver
Features of medullary carcinoma
- Neuroendocrine neoplasm derived from parafollicular C cells
- 80% sporadic - adults 5-6th decade
- 20% familial - MEN - younger patients
Features of anaplastic carcinoma
- Occur in elderly patients
- Very aggressive
- Metastases common
- Most cases death within one year due to local invasion
Functions of parathyroids
- Activity controlled by level of free calcium in blood
- Decreased calcium stimulates release of PTH
- PTH
–Activates osteoclasts
–Increases renal tubular reabsorption of calcium
–Increases conversion of vitamin D to its active form
–Increases urinary phosphate excretion
–Increases intestinal calcium absorption
Causes of hyperparathyroidism
- 80-90% - solitary adenoma
- 10-20% hyperplasia of all 4 glands
–Sporadic or component of MEN type 1
•<1% carcinoma
Causes of hypoparathyroidism
- Surgical ablation
- Congenital absence
- Autoimmune
Clinical manifestations of hypoparathyroidism
–Neuromuscular irritability - tingling, muscle spasms, tetany
–Cardiac arrhythmias
–Fits
–Cataracts

Adrenal gland
Zona glomerulosa secretes?
Aldosterone
Zona fasciculata secretes?
GCs
Zona reticularis secretes?
Androgens and GCs
Adrenal medulla secretes
Nadr + Adr
Manifestations of adrenocortical hyperfunction
- Cushing’s syndrome – excess glucocorticoids
- Hyperaldosteronism
- Virilising syndromes – excess androgens
Clinical features of Cushing’s syndrome
- Hypertension and weight gain
- Truncal obesity
- ‘moon’ facies
- ‘buffalo hump’
- Cutaneous striae
What is the most common cause of Cushing’s
Administration of exogenous GCs
Atrophic adrenal glands
What are the endogenous causes of Cushing’s syndrome
- >50% due to primary hypothalamic-pituitary disease with increased ACTH – Cushing’s disease
- Most associated with ACTH-producing adenoma in the pituitary
- Some have hyperplasia of ACTH secreting cells in pituitary
- Adrenal glands show nodular cortical hyperplasia
- 30% of cases – primary adrenal
- Most cases are due to a solitary neoplasm
–Adenoma
–Carcinoma
•Less commonly due to bilateral hyperplasia
What lung tumour associated with ectopic ACTH secretion
Small Cell Carcinoma
Adrenals show bilateral hyperplasia


Features of hyperaldosteronism
•Primary
–35 % aldosterone secreting adenoma – Conn’s syndrome
–60 % bilateral adrenal hyperplasia
–Clinical manifestations are hypertension and hypokalaemia
–Accounts for <1% of causes of hypertension but important to recognise as surgically correctable
Conn’s syndrome=
Primary hyperaldosteronism
Adrenogenital syndromes and neoplasms?
More commonly associated with carcinoma than adenoma
•Congenital adrenal hyperplasias
–Group of autosomal recessive disorders
–Hereditary defects in enzymes involved in cortisol biosynthesis
–Decreased cortisol results in increased ACTH, adrenal stimulation and increased androgen synthesis
–May present in childhood or less commonly in adults
Causes of adrenal insufficiency
Primary
Secondary due to reduced ACTH
Causes of acute primary adrenal insufficiency
–Sudden withdrawal of corticosteroid therapy
–Haemorrhage (neonates)
–Sepsis with DIC (Waterhouse-Friderichson syndrome)
Waterhouse-Friderichson syndrome
Waterhouse–Friderichsen syndrome (WFS), hemorrhagic adrenalitis or fulminant meningococcemia is defined as adrenal gland failure due to bleeding into the adrenal glands, commonly caused by severe bacterial infection: Typically the pathogen is the meningococcus Neisseria meningitidis.
Addison’s disease
Chronic primary adrenal insufficiency
Causes of chronic primary adrenal insufficiency
–Autoimmune (75-90%)
–TB
–HIV
–Metastatic tumour (lung and breast particularly)
–Rarely amyloid, fungal infections, haemochromatosis, sarcoidosis
What are the adrenocortical neoplasms
•Adenomas
–most non-functional
–May be associated with Cushing’s syndrome or Conn’s syndrome
•Carcinomas
–Rare
–Usually large
–More commonly associated with virilizing syndrome than adenoma
What are the neoplasms of the adrenal medulla
Phaeo
Neuroblastoma
Rule of 10s in phaeo?
–10% arise in association with a familial syndrome inc. MEN 2A and 2B, von Hippel-Lindau disease and Sturge-Weber syndrome
–10% are bilateral
–10% are malignant
–In addition 10% of catecholamine-secreting tumours arise outside the adrenal (paragangliomas)
A 65 year old man is in hospital after suffering an acute myocardial infarction. The house officer hears a pansystolic murmur on auscultation.
A.
Dilated cardiomyopathy
B.
Hypertrophic cardiomyopathy
C.
Myomalacia cordis
D.
Rheumatic fever
E.
Congenital septal defect
F.
Pericardial effusion
G.
Infective endocarditis
H.
Aortic stenosis
I.
Myxomatous mitral valve
J.
Pericarditis
K.
Mitral regurgitation
L.
Aortic regurgitation
Myomalacia cordis
A 46 year old women presents to A&E out of breath and with severe chest pain. On examination a mid systolic click late systolic murmur is revealed.
A.
Dilated cardiomyopathy
B.
Hypertrophic cardiomyopathy
C.
Myomalacia cordis
D.
Rheumatic fever
E.
Congenital septal defect
F.
Pericardial effusion
G.
Infective endocarditis
H.
Aortic stenosis
I.
Myxomatous mitral valve
J.
Pericarditis
K.
Mitral regurgitation
L.
Aortic regurgitation
Myxomatous mitral valve
Myomalacia cordis
Myomalacia cordis (weakness of cardiac muscle due to disintegration of dead myocytes):
Rupture of weakened cardiac muscle may result in:
· Mitral regurgitation:
Apical thrill, systolic murmur and pulmonary oedema
· Ventricular septal defect:
Left sternal edge thrill, murmur, hypotension
· External rupture:
Cardiac tamponade
A 68 year old smoker presents with jaundice and worsening abdominal and back pain. Scratch marks are seen on his arms and legs. He has lost 5kg in 2 months. Ultrasound shows dilated intrahepatic bile ducts.
A.
Carcinoma head of the pancreas
B.
Gallstones
C.
VIPoma (Werner Morrison syndrome)
D.
Haemochromatosis
E.
Vibrio cholerae infection
F.
Iatrogenic pancreatitis
G.
Insulinoma
H.
Renal tubular acidosis
I.
Gallstone pancreatitis
J.
Pseudocysts
K.
Hypercalcaemia
L.
Chronic alcoholic pancreatitis
M.
Cystic fibrosis
N.
Carcinoma tail of the pancreas
Carcinoma head of pancreas
What differentiates between carcinoma of the head of pancreas and tail of pancreas?
Carcinoma of the head causes jaundice, tail does not
A 59 year old widow complains of persistent back pain, loss of appetite and that she has dropped from dress size 18 to a size 14 in just 2 months. She was recently diagnosed with diabetes. A large central mass is palpable as well hepatosplenomegaly.
A.
Carcinoma head of the pancreas
B.
Gallstones
C.
VIPoma (Werner Morrison syndrome)
D.
Haemochromatosis
E.
Vibrio cholerae infection
F.
Iatrogenic pancreatitis
G.
Insulinoma
H.
Renal tubular acidosis
I.
Gallstone pancreatitis
J.
Pseudocysts
K.
Hypercalcaemia
L.
Chronic alcoholic pancreatitis
M.
Cystic fibrosis
N.
Carcinoma tail of the pancreas
Carcinoma tail of the pancreas
A 39 year old Nepalese man presents with severe watery diarrhoea. He is found to have hypokalaemia and, surprisingly, a metabolic acidosis. A RUQ mass is detected by contrast-enhanced spiral CT scanning. Stool bicarb is high and urine anion gap is negative.
A.
Carcinoma head of the pancreas
B.
Gallstones
C.
VIPoma (Werner Morrison syndrome)
D.
Haemochromatosis
E.
Vibrio cholerae infection
F.
Iatrogenic pancreatitis
G.
Insulinoma
H.
Renal tubular acidosis
I.
Gallstone pancreatitis
J.
Pseudocysts
K.
Hypercalcaemia
L.
Chronic alcoholic pancreatitis
M.
Cystic fibrosis
N.
Carcinoma tail of the pancreas
VIPoma
(Werner Morrison syndrome)
profound and chronic watery diarrhea and resultant dehydration, hypokalemia, achlorhydria, acidosis, vasodilation (flushing and hypotension), hypercalcemia and hyperglycemia.[3]
VIPoma
Werner Morrison Syndrome
Werner Morrison Syndrome
A VIPoma (also known as Verner–Morrison syndrome, after the physicians who first described it)[1] is a rare (1 per 10,000,000 per year) endocrine tumor,[2] usually (about 90%) originating from non-β islet cell of the pancreas, that produce vasoactive intestinal peptide (VIP). It may be associated with multiple endocrine neoplasia type 1.
The massive amounts of VIP in turn cause profound and chronic watery diarrhea and resultant dehydration, hypokalemia, achlorhydria (hence WDHA-syndrome, or pancreatic cholera syndrome), acidosis, vasodilation (flushing and hypotension), hypercalcemia and hyperglycemia.[3]
65 year old female with a large, cystic mass on tail of pancreas imaged using computed tomography. Further cytology reported the presence of epithelium
A.
Cystadenoma
B.
Carcinoma of the Pancreas
C.
Whipples’ resection
D.
Cystic Fibrosis
E.
Gall Bladder
F.
Agenesis
G.
Alcoholism
H.
Jaundice
I.
Pancreatitis
J.
Thrombophlebitis
K.
Trousseau’s Syndrome
L.
Scorpion Sting
M.
Hyperlipidaemia
N.
Type 1 Diabetes
O.
Pancreas Divisum
P.
Pseudocyst
Cystadenoma
55 year old, diabetic, afro-Caribbean male presents with weight loss, poor diet and a gnawing pain in his back, which is sometimes felt ‘under his chest’
A.
Cystadenoma
B.
Carcinoma of the Pancreas
C.
Whipples’ resection
D.
Cystic Fibrosis
E.
Gall Bladder
F.
Agenesis
G.
Alcoholism
H.
Jaundice
I.
Pancreatitis
J.
Thrombophlebitis
K.
Trousseau’s Syndrome
L.
Scorpion Sting
M.
Hyperlipidaemia
N.
Type 1 Diabetes
O.
Pancreas Divisum
P.
Pseudocyst
Carcinoma of the Pancreas
What differentiates between a cystadenoma and a pancreatic pseduocyst?
Pancreatic pseudocyst is a fluid filled collection contained within a well-defined capsule of fibrous or granulation tissue or a combination of both. It does not possess an epithelial lining. A cystadenoma has an epithelial wall or capsule that contains a fluid collection.
Caused by the action of acid and pepsin on the duodenal mucosa. Associated with increased output of stomach acid. Symptoms include pain in the upper abdomen, especially when the stomach is empty.
A.
Oesophageal varices
B.
Duodenal ulceration
C.
Pernicious anaemia
D.
Intestinal metaplasia
E.
Reflux oesophagitis
F.
Campylobacter jejuni
G.
Gastric cancer
H.
Helicobacter pylori
I.
Gastric ulcer
J.
Squamous carcinoma
K.
Barrett’s oesophagus
L.
Adenocarcinoma
Duodenal ulceration
Around 10 % eventually get primary lymphoma (less often, carcinoma) of the gut if not properly treated. HLA B8 is linked with this
A.
Pernicious anaemia
B.
H. pylori infection
C.
Coeliac disease
D.
Intestinal metaplasia
E.
Normal oesophagus
F.
GORD
G.
Chronic gastritis
H.
Partial villus atrophy
I.
Normal stomach
J.
Peptic ulcer
K.
Barrett’s oesophagus
Coeliac
A 30 year old female complaining of diarhorrea and weight loss. Biopsy of duodenum shows increased intraepithelial cytotoxic T cells.
A.
Acute Gastritis
B.
Pernicious Anaemia
C.
Barretts Oesophagus
D.
Oesophageal Adenocarcinoma
E.
Gastric Carcinoma
F.
GORD
G.
Coeliac Disease
H.
Oesophageal Varices
I.
Active Chronic Gastritis
J.
Duodenal Ulcer
Coeliac
A 26 year old man presents with watery diarrhoea, abdominal cramps, nausea, vomiting and a low grade fever. It started 3 days after eating some undercooked meat at a barbecue.
.
Carcinoma of the oesophagus
B.
Whipple’s disease
C.
Coeliac disease
D.
Cryptosporidiosis
E.
Partial villous atrophy
F.
Hiatus hernia
G.
Helicobacter pylori
H.
Gastric ulcer
I.
Gastro-oesophageal disease
J.
Barrett’s oesophagus
K.
Microsporidiosis
L.
Mucosal associated lymphoid tumour
M.
Tropical sprue
N.
Pernicious anaemia
O.
Lymphoma
P.
Duodenal ulcer
Cryptosporidios
A 58 year old female presents with malnutrition. She complains of abdominal pain, weight loss and arthritis. She has steatorrhoea. A jejunal biopsy showed periodic acid-Schiff (PAS)-positive macrophages
A.
Carcinoma of the oesophagus
B.
Whipple’s disease
C.
Coeliac disease
D.
Cryptosporidiosis
E.
Partial villous atrophy
F.
Hiatus hernia
G.
Helicobacter pylori
H.
Gastric ulcer
I.
Gastro-oesophageal disease
J.
Barrett’s oesophagus
K.
Microsporidiosis
L.
Mucosal associated lymphoid tumour
M.
Tropical sprue
N.
Pernicious anaemia
O.
Lymphoma
P.
Duodenal ulcer
Whipple’s disease
A 20-year-old student gives an 8 hour history of very frequent vomiting and epigastric cramping. O/E she is pale and shivering. Her serum WBC is normal.
A.
Gastroenteritis (Staphylococcus aureus)
B.
Gastric ulcer
C.
Mucosal-associated lymphoid tumour
D.
Gastro-oesophageal reflux disease
E.
Bulbar palsy
F.
Zollinger-Ellison syndrome
G.
Duodenal ulcer
H.
Adenocarcinoma
I.
Barrett’s oesophagus
J.
Mallory-Weiss tear
K.
Gastroenteritis (Salmonella)
L.
Haemorrhagic gastritis
M.
Pyloric stenosis
N.
Achalasia
Gastroenteritis- staph
How to classify causes of splenomegaly?
Size:
Massive, moderate, mild
Causes of massive splenomegaly
CML, myelofibrosis (UK)
Malaria, Kala-Azar (worldwide)
Causes of moderate splenomegaly
Portal HTN
Lymphoma
CLL
Thalassaemia
Metabolic diseases e.g. Gaucher’s
Causes of mild splenomegaly
Infection e.g. EBV, hepatitis, bacterial
CTD e.g. RA, PAN, SLE, Felty’s
Infiltrative disorders: amyloidosis, sarcoidosis
AA amyloidosis
Secondary to inflammatory conditions e.g. RA, Crohn’s etc
Pancreatic pseudocysts usually develop
Post surgery
Intestinal metaplasia
Intestinal metaplasia describes the transformation of an epithelium to the columnar epithelium found in the intestines. The distinguishing feature of intestinal epithelium is the presence of goblet cells. (NB. the stomach epithelium is also columnar, but it doesn’t contain the distinctive goblet cells. It contains foveolar cells which form glands and secrete mucin) Intestinal metaplasia is a response to chronic inflammation. It is commonly associated with chronic gastritis - H. pylori-induced or autoimmune. The presence of intestinal metaplasia is a strong risk-factor for the development of adenocarcinoma.
Barret’s oesophagus
Barrett’s oesophagus is another example of intestinal metaplasia due to chronic inflammation. This time chronic GORD is the cause. Again, the intestinal metaplasia is strongly associated with the development of adenocarcinoma. GORD and Barrett’s is the reason that oesophageal adenocarinoma tends to affect the distal third of the oesophagus while squamus cell oesophageal cancer affects the upper two thirds.
Which HLA alleles are assocaited with Coeliac disease?
DQ2 most strongly
DQ8
What differentiates between acute and chronic gastritis
Chronic gastritis is usually caused by H. pylori which would result in an antral gastritis. This patient had erosions throughout the gastric mucosa. Chronic gastritis would be have lymphocytes and plasma cells on biopsy - this patient had a neutrophilic infiltrate which is typical of an acute gastritis. Focal, acute mucosal defects are a well-known complication of NSAIDs which this patient was probably taking.
Oesophageal carcinoma tissue type lower
Adenocarcinoma
Oesphageal carcinoma tissue type middle
Squamous cell carcinoma
A 20-year-old student gives an 8 hour history of very frequent vomiting and epigastric cramping. O/E she is pale and shivering. Her serum WBC is normal.
Why is the WBC normal?
Staphylococcus aureus is a leading cause of gastroenteritis resulting from the consumption of contaminated food. Staphylococcal food poisoning is due to the absorption of staphylococcal enterotoxins preformed in the food. The onset of symptoms is rapid (from 30 min to 8 h) and usually spontaneous remission is observed after 24 h. As the presentation is so acute, the WCC often have not had a chance to increase (lag-time). The symptoms of staphylococcal food poisoning are abdominal cramps, nausea, vomiting, sometimes followed by diarrhea (never diarrhea alone).
A 26 year old gentleman presents to A and E with acute abdominal pain. He has lost 5 kg of weight in the last 2 weeks, and has been passing bloody diarrhoea with mucus. He is pyrexial and on examination is noted to have angular stomatitis.
A.
Diverticulitis
B.
Diverticular disease
C.
Angiodysplasia
D.
Crohn’s disease
E.
Intestinal TB
F.
Ulcerative colitis
G.
Gastroenteritis
H.
Colorectal carcinoma
I.
Pseudomembranous colitis
J.
Tubular adenoma
K.
Ischaemic colitis
L.
Sigmoid volvulus
Crohn’s disease
A 62 year old housewife returns to your outpatient clinic following another incidence of the passing of blood. Previous sigmoidoscopy, DRE and barium enema’s have failed to identify any lesion and she denies weight loss and diarrhoea. However, blood tests show a microcytic anaemia.
A.
Appendicitis
B.
Anorectal abscess
C.
Angiodysplasia
D.
Anterior rectocoele
E.
Diverticular disease
F.
Adenocarcinoma Duke’s stage B
G.
Diverticulitis
H.
Perianal haematoma
I.
Benign adenoma
J.
Haemorrhoids
K.
Villous adenoma
L.
Crohn’s disease
M.
Ischaemic colitis
N.
Irritable bowel syndrome
O.
Adenocarcinoma Duke’s stage C1
P.
Ulcerative colitis
olonic angiodysplasia is a common cause of acute or chronic rectal bleeding and iron deficiency anaemia. Angiodysplasias are tiny - 1-5 mm in diameter - hamartomatous capillary lesions in the colonic wall which produce bleeding out of proportion to their size. They are believed to be acquired, possibly as a result of tension on the veins where they pass through the muscularis. Diagnosis: subtraction mesenteric arteriography may demonstrate bleeding if rapid colonscopy: may visualise lesion Treatment: electrical coagulation via the colonoscope resection of segment of colon if the above is unsuccessful
A 73 year old woman attends complaining of recent onset of tiredness. She is pale and has hepatosplenomegaly and generalised lymphadenopathy in the neck, axillae and groins.
A.
Sarcoidosis
B.
Severe emphysema
C.
Chronic lymphocytic leukaemia
D.
Cirrhosis with hepatoma
E.
Hameochromatosis
F.
Liver abscess
G.
Gaucher’s disease
H.
Portal vein thrombosis
I.
Congestive heart failure
J.
Polycythaemia rubra vera
K.
Chronic myeloid leukaemia
Chronic lymphocytic leukaemia
A 45 year old male was admitted to hospital after having a fall. Investigations reveal a leucocytosis with elevated bilirubin and transferases. Levels were albumin, folate and vitamin B12 were low. Clotting studies showed a prolonged prothrombin time.
.
Hereditary haemochromatosis
B.
Alcoholic liver disease
C.
Hepatitis A
D.
Hepatitis B
E.
Hepatocellular carcinoma
F.
Wilson’s disease
G.
Autoimmune hepatitis
H.
Primary biliary cirrhosis
Alcoholic liver disease
A 35 year old patient with known Hepatitis B visits the doctor complaining of recent weight loss, loss of appetite, fevers and an ache in the right hypochondrium. On examination an enlarged, irregular, tender liver can be palpated.
A.
Primary sclerosing cholangitis
B.
Alpha1 antitrypsin deficiency
C.
Wilson’s disease
D.
Hereditary haemochromatosis
E.
Hepatocellular carcinoma
F.
Hepatitis B
G.
Primary biliary cirrhosis
H.
Ascending cholangitis
I.
Budd-Chiari syndrome
J.
Hepatitis C
K.
Alcoholic hepatitis
Hepatocellular carcinoma
A 65 year old retired factory worker presents with dysuria and frequency. He has also noticed that his urine is red. PSA is normal.
A.
Renal cell carcinoma
B.
Pyelonephritis
C.
Cystitis
D.
Urothelial carcinoma
E.
Adult PCKD
F.
Bladder cancer
G.
Prostatitis
H.
Prostate cancer
Bladder cancer
54 year old man presents to A&E with fever and weakness. Blood tests show hypercalcaemia . On eaxamination a palpable mass is found in the right loin area
A.
Renal cell carcinoma
B.
Pyelonephritis
C.
Cystitis
D.
Urothelial carcinoma
E.
Adult PCKD
F.
Bladder cancer
G.
Prostatitis
H.
Prostate cancer
.
Renal cell carcinoma
A 30 year old woman presents with lower abdominal pain accompanied by fever. She has been using an intra uterine contraceptive device since her wedding three years ago.
A.
Herpes virus infection
B.
Endometriosis
C.
Fibroids
D.
Endometritis
E.
Ectopic pregnancy
F.
Cervical polyps
G.
Arias-Stella phenomenon
H.
Polycystic ovary syndrome
I.
Candidiasis
J.
Salpingitis
Endometritis
A 30 year old woman attends a fertility clinic as her and her husband are unable to conceive. For the last few years her periods have been fairly heavy and prolonged. She has stopped wearing skirts because she thinks her ankles look fat.
A.
Herpes virus infection
B.
Endometriosis
C.
Fibroids
D.
Endometritis
E.
Ectopic pregnancy
F.
Cervical polyps
G.
Arias-Stella phenomenon
H.
Polycystic ovary syndrome
I.
Candidiasis
J.
Salpingitis
Fibroids
What is Charcot’s triad
Fever
RUQ pain
Jaundice
with rigors etc
Bacterial cholangitis
Features of PSC
Primary sclerosing cholangitis is a rare disease of unknown aetiology characterised by chronic inflammation and fibrosis of the bile duct. - usually men - associated with IBD, particularly ulcerative colitis - may be complicated by strictures, cholangitis, cholangiocarcinoma - autoantibodies are less frequent than in autoimmune chronic active hepatitis and primary biliary cirrhosis: in an exam, ANCA positivity might be a clue as P-ANCA positive in more than 50% of cases. Antimitochondrial antibodies almost never occur. - Liver Bx: fibrous obliterating cholangitis loss of interlobular and adjacent septal bile ducts - ERCP shows multiple annular strictures, separated by round or slightly dilated duct segments and the intrahepatic and extrahepatic bile ducts have a beaded appearance
fibrous obliterating cholangitis loss of interlobular and adjacent septal bile ducts - ERCP shows multiple annular strictures, separated by round or slightly dilated duct segments and the intrahepatic and extrahepatic bile ducts have a beaded appearance
PSC
Features of PBC
Primary biliary cirrhosis is a chronic, progressive, cholestatic liver disease which classically affects middle aged women (rare under 30 yrs). 95% of patients have anti-mitochondrial autoantibodies, especially E2 component of the pyruvate dehydrogenase complex. Pruritus and fatigue common, but also remember xanthelasma, hyperlipidaemia, feats of chronic liver disease.
A 40 year old woman has always known cramping pain associated with her periods – which have usually been heavy. Recently this pain has become constant throughout the month, and her periods have become more frequent. She claims never to have used oral contraception and has no children. She is abstaining from sexual intercourse as it is too painful.
Although you may classically think of the pain of endometriosis as cyclical - responding to the hormonal influence of menstruation, the history is very variable and may be progressively worse to even continuous pelvic pain. Constant pain is often due to adhesions (which this patient may have now developed). In the history - nulliparity, due to subfertility secondary to the endometriosis itself or abstinence due to deep dyspareunia is common. Rare, but possible…are symptoms such as cyclical haematuria or haemopytsis….(which is important to keep in your differential diagnosis, albeit at the bottom of your list…) or painful, cyclical, expanding masses in a pelvic scar.
This type of bone is 80-90% calcified and its function is mainly mechanical and protective.
Cortical
This type of bone is immature and usually pathological.
A.
Lamellar
B.
Woven
C.
Osteoblast
D.
Osteoclast
E.
Metaphysis
F.
Osteocyte
G.
Periosteum
H.
Diaphysis
I.
Cortical
J.
Endosteum
K.
Epiphysis
L.
Cancellous
Woven
A 15-year-old male presents with a 2-month history of increasing pain in his right upper arm. Bone biopsy reveals sheets of cells with small, primitive nuclei and scanty cytoplasm. A positive immunoreactivity is seen with the MIC2 (CD99) antibody.
A.
Primary osteosarcoma
B.
Osteogenesis imperfecta
C.
Osteopetrosis
D.
Osteomalacia
E.
Osteomyelitis
F.
Osteoporosis
G.
Osteoarthritis
H.
Chondrosarcoma
I.
Ewing’s sarcoma
Ewing’s sarcoma
A 20-year-old male complains of a progressively enlarging painful mass on his right upper arm. Radiology demonstrates a lytic lesion of the proximal humerus with an accompanying Codman triangle. Microscopically, pleomorphic mesenchymal cells producing dark-staining osteoid are seen.
A.
Primary osteosarcoma
B.
Osteogenesis imperfecta
C.
Osteopetrosis
D.
Osteomalacia
E.
Osteomyelitis
F.
Osteoporosis
G.
Osteoarthritis
H.
Chondrosarcoma
I.
Ewing’s sarcoma
Primary osteosarcoma
A 24 year old police woman attends the clinic as her GP suspects she may have a parathyroid tumour. She has raised PTH and serum calcium. After a 24hr urinary collection it is noted the patient has a low urine calcium output
A.
Osteitis fibrosa
B.
Secondary hyperparathyroidism with chronic renal osteodystrophy
C.
Paget’s disease
D.
Cushing’s syndrome
E.
Cushing’s disease
F.
Familial hypocalcuric hypercalcaemia
G.
Primary hyperparathyroidism
H.
Tertiary hyperparathyroidism
I.
Osteomalacia
J.
Osteoporosis
K.
Primary hypthyroidism (myxoedema)
Familial hypocalcuric hypercalcaemia
ou see a new patient for the first time at the surgery, a 14 year old boy, complaining of pain in his right leg for 9 months and contracture of the right knee which has developed over this period. The child looks stunted and you can see bowing of the lower extremities in ambulators. His notes have not arrived yet from his previous doctor but his mother tells you ‘he was born 3 weeks premature with two small kidneys and three failed transplants means he has dialysis four times a day’. His blood results later show a raised PTH and phostate, low calcium and 1,25(OH)vitamin D. He is also acidotic.
A.
Osteitis fibrosa
B.
Secondary hyperparathyroidism with chronic renal osteodystrophy
C.
Paget’s disease
D.
Cushing’s syndrome
E.
Cushing’s disease
F.
Familial hypocalcuric hypercalcaemia
G.
Primary hyperparathyroidism
H.
Tertiary hyperparathyroidism
I.
Osteomalacia
J.
Osteoporosis
K.
Primary hypthyroidism (myxoedema)
Secondary hyperparathyroidism with chronic renal osteodystrophy
An 18 year old student presents to his GP with focal pain in his left fore-arm which is tender to touch and worsens at night. The pain is relieved with aspirin. An X-ray shows a 1cm are of radio-lucency in the tibia surrounded by dense bone.
A.
Osteoclastoma
B.
Osteosarcoma
C.
Osteochondroma
D.
Chondrosarcoma
E.
Ewing’s tumour
F.
Osteoid osteoma
G.
Osteoporosis
H.
Simple bone cyst
I.
Osteitis
J.
Trauma
K.
Rheumatoid arthritis
L.
Osteoarthritis
M.
Metastases
N.
Fibrous dysplasia
O.
Echondroma
Osteoid osteoma
A 14 year old boy complains to you of a painless lump on his left thigh, just above the knee which is slowly growing. His past medical history reveals that he fractured his femur in the same location several years before.
A.
Osteoclastoma
B.
Osteosarcoma
C.
Osteochondroma
D.
Chondrosarcoma
E.
Ewing’s tumour
F.
Osteoid osteoma
G.
Osteoporosis
H.
Simple bone cyst
I.
Osteitis
J.
Trauma
K.
Rheumatoid arthritis
L.
Osteoarthritis
M.
Metastases
N.
Fibrous dysplasia
O.
Echondroma
Osteochondroma
A 15 year old girl shows you a small lump on her upper arm on routine examination. She says the lump has been present for a couple of years and has slowly moved down, away from her shoulder.
A.
Osteoclastoma
B.
Osteosarcoma
C.
Osteochondroma
D.
Chondrosarcoma
E.
Ewing’s tumour
F.
Osteoid osteoma
G.
Osteoporosis
H.
Simple bone cyst
I.
Osteitis
J.
Trauma
K.
Rheumatoid arthritis
L.
Osteoarthritis
M.
Metastases
N.
Fibrous dysplasia
O.
Echondroma
Simple bone cyst
A 50 year old lady presents with pain in her jaw. She suffers from Paget’s disease. A ‘sunburst’ appearance is seen on X-ray along with a lifted periosteum (Codman’s triangle).
A.
Osteoclastoma
B.
Osteosarcoma
C.
Osteochondroma
D.
Chondrosarcoma
E.
Ewing’s tumour
F.
Osteoid osteoma
G.
Osteoporosis
H.
Simple bone cyst
I.
Osteitis
J.
Trauma
K.
Rheumatoid arthritis
L.
Osteoarthritis
M.
Metastases
N.
Fibrous dysplasia
O.
Echondroma
Osteosarcoma
An 8 year old boy is brought to his GP by his parents with pain in his hips and a fever. Blood results demonstrate a raised ESR and biopsy histology shows droplets of glycogen in the cytoplasm of small round cells in the pelvic bones.
A.
Osteoclastoma
B.
Osteosarcoma
C.
Osteochondroma
D.
Chondrosarcoma
E.
Ewing’s tumour
F.
Osteoid osteoma
G.
Osteoporosis
H.
Simple bone cyst
I.
Osteitis
J.
Trauma
K.
Rheumatoid arthritis
L.
Osteoarthritis
M.
Metastases
N.
Fibrous dysplasia
O.
Echondroma
Ewing’s tumour
70 year old man presents to his GP with a four day history of haemoptysis. He has noticed he has been loosing weight over the last 4 months and has felt tired and unwell. On examination he has bilateral ptosis and proximal weakness in the limbs which improves on repeated testing
A.
Cystic fibrosis
B.
Acute asthma
C.
Cryptogenic fibrosing alveolitis
D.
Tuberculosis
E.
Emphysema
F.
Pulmonary oedema
G.
Pulmonary embolism
H.
Squamous cell carcinoma
I.
Pneumococcal pneumonia
J.
Small cell carcinoma
K.
Thymoma
L.
Chronic bronchitis
M.
Mycoplasma pneumonia
Small cell carcinoma
A 35 year old woman presents with weight loss and tiredness. Her GP examines her and finds that she also has a fine tremor and is sweaty. Investigations: TSH <0.01, Free T4 36.0. There is low uptake on a technetium scan. The aetiology could be viral or autoimmune.
A.
Grave’s disease
B.
De Quervain’s
C.
Hashimoto’s thyroiditis
D.
Euthyroid state
E.
Medullary carcinoma
F.
Thyroid storm
G.
Subacute thyroiditis
H.
Postpartum thyroiditis
I.
Papillary carcinoma
Subacute thyroiditis
An 18 year old man notices a lump on his neck and goes to his GP. As well as the lump, the GP discovers cervical lymphadenopathy. There is no family history of any endocrine disorder, nor is he suffering from any other illness. Thyroglobulin is 140.
A.
Grave’s disease
B.
De Quervain’s
C.
Hashimoto’s thyroiditis
D.
Euthyroid state
E.
Medullary carcinoma
F.
Thyroid storm
G.
Subacute thyroiditis
H.
Postpartum thyroiditis
I.
Papillary carcinoma
Papillary carcinoma
eoplasms found in women aged between 30 and 40 as ovarian masses, usually unilateral. They are usually benign (90%) and are often the largest ovarian neoplasm.
A.
Cervical Squamous Cell Carcinoma
B.
Cervical Intraepithelial Neoplasia (CIN)
C.
Endometrial Adenocarcinoma
D.
Endometrial Polyps
E.
Vaginal Squamous Cell Carcinoma
F.
Mucinous Tumour
G.
Serous Tumour
H.
Teratoma (Mature)
I.
Endometrioid Tumour
J.
Teratoma (Immature)
Mucinous ovarian neoplasms account for 10-15% of all ovarian tumours and are rarely malignant (~10-15%). They are rarely bilateral, but can be very large. In contrast, whilst serous tumours are also usually benign (30% malignant) and common - accounting for 30% of all ovarian tumours - they have a strong tendency to be bilateral.
Squamous epithelium mixed with intestinal epithelium
A.
Serous cystadenocarcinoma
B.
Mature cystic teratoma
C.
Thecoma
D.
Dysgerminoma
E.
Fibroma
F.
Krukenberg tumour
G.
Mucinous cystadenocarcinoma
H.
Clear cell tumour
I.
Sertoli-Leydig tumour
J.
Immature teratoma
K.
Granulosa cell tumours
Mature cystic teratoma
Fibrous tissue containing spindle cells and lipid
A.
Serous cystadenocarcinoma
B.
Mature cystic teratoma
C.
Thecoma
D.
Dysgerminoma
E.
Fibroma
F.
Krukenberg tumour
G.
Mucinous cystadenocarcinoma
H.
Clear cell tumour
I.
Sertoli-Leydig tumour
J.
Immature teratoma
K.
Granulosa cell tumours
.Thecoma
Feedback:
In a fibroma, there are intersecting bundles of spindle cells. Thecomas in addition contain lipid.
Malignant cells surrounded by serous fluid and Psammoma bodies
A.
Serous cystadenocarcinoma
B.
Mature cystic teratoma
C.
Thecoma
D.
Dysgerminoma
E.
Fibroma
F.
Krukenberg tumour
G.
Mucinous cystadenocarcinoma
H.
Clear cell tumour
I.
Sertoli-Leydig tumour
J.
Immature teratoma
K.
Granulosa cell tumours
Serous cystadenocarcinoma
Germ cells mixed with lymphocytes
A.
Serous cystadenocarcinoma
B.
Mature cystic teratoma
C.
Thecoma
D.
Dysgerminoma
E.
Fibroma
F.
Krukenberg tumour
G.
Mucinous cystadenocarcinoma
H.
Clear cell tumour
I.
Sertoli-Leydig tumour
J.
Immature teratoma
K.
Granulosa cell tumours
Dysgerminoma
Seminomatous germ cell tumours are
radioSensitive, present in the 30s ; 15% secrete HCG ; 0% secrete AFP.
radioSensitive, present in the 30s ; 15% secrete HCG ; 0% secrete AFP.
Seminomatous germ cell tumours
Nonseminatous GCTs are
present in the 20s, and may secrete AFP and/or HCG.
Germ cell tumour originating in testis that is radiosensitive and classically presents in the 4th decade.
A.
Diffuse large B cell lymphoma (DLBCL)
B.
Seminoma
C.
Embryonal carcinoma
D.
Acute lymphoblastic leukaemia/lymphoma (ALL)
E.
Choriocarcinoma
F.
Teratoma
G.
Leydig cell tumour
H.
Acute myeloid leukaemia (AML)
I.
Sertoli cell tumour
J.
Yolk sac (endodermal sinus) tumour
Seminoma
Very aggressive tumour producing HCG and AFP; neoplastic cells are anaplastic.
A.
Diffuse large B cell lymphoma (DLBCL)
B.
Seminoma
C.
Embryonal carcinoma
D.
Acute lymphoblastic leukaemia/lymphoma (ALL)
E.
Choriocarcinoma
F.
Teratoma
G.
Leydig cell tumour
H.
Acute myeloid leukaemia (AML)
I.
Sertoli cell tumour
J.
Yolk sac (endodermal sinus) tumour
Embryonal carcinoma
Very aggressive HCG-producing tumour composed of cytotrophoblast and syncytiotrophoblast cells that metastasizes early.
A.
Diffuse large B cell lymphoma (DLBCL)
B.
Seminoma
C.
Embryonal carcinoma
D.
Acute lymphoblastic leukaemia/lymphoma (ALL)
E.
Choriocarcinoma
F.
Teratoma
G.
Leydig cell tumour
H.
Acute myeloid leukaemia (AML)
I.
Sertoli cell tumour
J.
Yolk sac (endodermal sinus) tumour
Choriocarcinoma
Commonest malignant cause of testicular mass in those aged under 5..
A.
Diffuse large B cell lymphoma (DLBCL)
B.
Seminoma
C.
Embryonal carcinoma
D.
Acute lymphoblastic leukaemia/lymphoma (ALL)
E.
Choriocarcinoma
F.
Teratoma
G.
Leydig cell tumour
H.
Acute myeloid leukaemia (AML)
I.
Sertoli cell tumour
J.
Yolk sac (endodermal sinus) tumour
Acute lymphoblastic leukaemia/lymphoma (ALL)
Commonest malignant cause of testicular mass in those aged 60.
A.
Diffuse large B cell lymphoma (DLBCL)
B.
Seminoma
C.
Embryonal carcinoma
D.
Acute lymphoblastic leukaemia/lymphoma (ALL)
E.
Choriocarcinoma
F.
Teratoma
G.
Leydig cell tumour
H.
Acute myeloid leukaemia (AML)
I.
Sertoli cell tumour
J.
Yolk sac (endodermal sinus) tumour
Diffuse large B cell lymphoma (DLBCL)
A 22-year-old lady presented with a vaginal discharge. Gram staining revealed “Clue cells” surrounded by rods, that were “Gram variable”.
A.
Neisseria gonorrheae
B.
Trichomonas vaginalis
C.
Herpes zoster
D.
Cervical Intraepithelial Neoplasia I
E.
Herpes simplex
F.
Cervical Intraepithelial Neoplasia III
G.
Cervical Intraepithelial Neoplasia II
H.
Granuloma Inguinale
I.
Candida albicans
J.
Group B Streptococcus
K.
Chlamydia
L.
Lymphogranuloma venereum
M.
Treponema pallidum (syphilis)
N.
Cervical polyps
O.
Gardnerella vaginalis
P.
Cervical Microglandular Hyperplasia
Gardnerella vaginalis
A pap smear taken from a chronic granulomatous ulcer shows a necrotic centre, periarteritis and endarteritis obliterans and an intense peripheral cellular infiltrate consisting mainly of mononuclear cells and giant cells.
A.
Neisseria gonorrheae
B.
Trichomonas vaginalis
C.
Herpes zoster
D.
Cervical Intraepithelial Neoplasia I
E.
Herpes simplex
F.
Cervical Intraepithelial Neoplasia III
G.
Cervical Intraepithelial Neoplasia II
H.
Granuloma Inguinale
I.
Candida albicans
J.
Group B Streptococcus
K.
Chlamydia
L.
Lymphogranuloma venereum
M.
Treponema pallidum (syphilis)
N.
Cervical polyps
O.
Gardnerella vaginalis
P.
Cervical Microglandular Hyperplasia
Treponema pallidum (syphilis)
A young lady is found to have chronic irritation and inflammation of the vulva. A pap smear and use of a silver stain reveals fungi within the keratin layer and superficial epithelium.
A.
Neisseria gonorrheae
B.
Trichomonas vaginalis
C.
Herpes zoster
D.
Cervical Intraepithelial Neoplasia I
E.
Herpes simplex
F.
Cervical Intraepithelial Neoplasia III
G.
Cervical Intraepithelial Neoplasia II
H.
Granuloma Inguinale
I.
Candida albicans
J.
Group B Streptococcus
K.
Chlamydia
L.
Lymphogranuloma venereum
M.
Treponema pallidum (syphilis)
N.
Cervical polyps
O.
Gardnerella vaginalis
P.
Cervical Microglandular Hyperplasia
Candida albicans
The metaplasia that occurs in the transformation zone involves which cell-types?
Glandular to squamous epithelium
A 27 year old woman undergoes a routine Pap test. Histology shows increased squamous epithelium with atypical cells showing koilocytosis (nuclear enlargement with perinuclear halo = clear area around nucleus). Follow-up colposcopy shows hyperchromatic nuclei present in the lower 1/3 of the epithelial layer from the basement membrane.
A.
Invasive cervical cancer
B.
CIN I
C.
Squamous to glandular epitheliem
D.
50-60 years
E.
CIN III
F.
45 years
G.
Glandular to squamous epithelium
H.
Ovarian cancer
I.
65 years
J.
30 - 40 years
K.
CIN II
CIN I
22 year old lady presented for her routine smear test. The test showed a slight increase in DNA staining, and also a slightly larger variation in size of nuclei. Further questioning revealed she was a single mother living with her boyfriend, and had had multiple sexual partners since her first sexual contact at age 16.
A.
Group B Streptococcus
B.
Cervical Microglandular Hyperplasia
C.
Lymphogranuloma venereum
D.
Cervical Intraepithelial Neoplasia III
E.
Trichomonas vaginalis
F.
Neisseria gonorrheae
G.
Cervical Intraepithelial Neoplasia I
H.
Mild dyskaryosis
I.
Herpes simplex
J.
Granuloma Inguinale
K.
Gardnerella vaginalis
L.
Treponema pallidum (syphilis)
M.
Severe dyskaryosis
N.
Chlamydia
O.
Candida albicans
Mild dyskaryosis
Single most useful first-line cytological investigation for the confirmation of the benign status of an ovarian cyst.
A.
Exfoliative (brush) cytology
B.
Cystoscopy
C.
Cone biopsy
D.
Endometrial tissue sampling
E.
Percutaneous FNA biopsy
F.
Stereotactic radiographic cytological sampling method
G.
Fine needle aspirate
H.
Core biopsy
I.
Fluid cytology of alveolar washings
Core biopsies give HISTOLOGY Fine needle aspirates give CYTOLOGY - cannot assess basement membrane penetration (ie invasion)
FNA
Has a 100% positive predictive value for malignant cytopathological diagnosis.
FNA
What is the importance of measuriny urinary calcium in ?hyperPTHism
Familial hypocalciuric hypercalcaemia (FHH) is a dominantly inherited condition caused by a mutation of the calcium sensing receptor. The result is that the kidney reabsorbes calcium very avidly as a primary problem. This results in hypocalciuria and hypercalcaemia. Because the parathyroid gland calcium receptor also does not work, the parathyroids continue to secrete excess PTH despite the high calcium, so it looks just like primary hyperparathyroidism. However removing the parathyroids does not help the calcium, as the primary cause of trouble is in the kidney. In true primary hyperparathyroidism however, the calcium that is filtered hugely exceeds the tubular maximum for calcium, so there is no chance of reabsorption, so the urinary calcium is high, with resulting renal stones (which NEVER happens in FHH). There are many surgeons who have operated wrongly on FHH patients, removing the parathyroid only to find the calcium does not get better. As a consequence, we have written guidelines to surgeons, reminding them of the importance of measuring urine calcium in such a patient before removing the parathyroid gland.
70 year old man presents to his GP with a four day history of haemoptysis. He has noticed he has been loosing weight over the last 4 months and has felt tired and unwell. On examination he has bilateral ptosis and proximal weakness in the limbs which improves on repeated testing — Correct answer: Small cell carcinoma why is this Small Cell Carcinoma?
This guy has Myasthenic Syndrome Eaton-Lambert syndrome which occurs in ~3% of people with small cell l ung cancer. In contrast with myasthenia gravis, the fatigue gets BETTER with repeated use of the muscle.
What differentiates between autoimmune and De Quervain’s thyroiditis
Autoimmune tend to be painless
endarteritis obliterans in the context of STIs=
Treponema pallidum
An 86 year old man is admitted to A&E having collapsed at his home. He is unconscious and a couple of days later he is still deeply unconscious. His pupils are pin-point and their reaction to light is difficult to see clearly
A.
Vascular dementia
B.
Cerebellar stroke
C.
Opiate overdose
D.
Transient Ischaemic attack
E.
Cerebral embolus
F.
Subarachnoid haemorrhage
G.
Panic attack
H.
Pontine haemorrhage
I.
Subdural haemorrhage
J.
Temporal arteritis
K.
Transient hypotension
L.
Brain stem infarction
M.
Extradural haemorrhage
N.
Raised intracranial pressure
O.
Alzheimer’s disease
P.
Meningitis
Brain stem infarction
Occlusion of this cerebral vessel can cause weakness and numbness in the contralateral lower limb and similar but milder symptoms in the contralateral upper limb.
A.
Anterior cerebral artery
B.
Lacunar infarcts
C.
Middle cerebral artery
D.
Berry’s aneurysm
E.
Vertebrobasilar circulation
F.
Extradural haemorrhage
G.
Bacterial meningitis
H.
Hydrocephalus
I.
Amyloidosis
J.
Viral meningitis
K.
Subarachnoid haemorrhage
L.
Posterior cerebral artery
M.
Stroke
N.
Transient ischaemic attack (TIA)
ACA
Transtentorial herniations can potentially compromise the sufficiency this particular part of the cerebral circulation and cause occipital lobe infarction.
A.
Anterior cerebral artery
B.
Lacunar infarcts
C.
Middle cerebral artery
D.
Berry’s aneurysm
E.
Vertebrobasilar circulation
F.
Extradural haemorrhage
G.
Bacterial meningitis
H.
Hydrocephalus
I.
Amyloidosis
J.
Viral meningitis
K.
Subarachnoid haemorrhage
L.
Posterior cerebral artery
M.
Stroke
N.
Transient ischaemic attack (TIA)
PCA
5 year old obese woman goes to GP about worsening headaches. They are especially bad in the mornings and she also feels nauseous with some visual disturbances.
A.
Subdural haemorrhage
B.
Trigeminal neuralgia
C.
Glaucoma
D.
Migraine
E.
Berry aneurysm
F.
Subarachnoid haemorrhage
G.
Tension headache
H.
Sinusitis
I.
Benign intracranial hypertension
BIH
An elderly diabetic lady with an acute headache and associated blurred vision and vomiting. Pain is localised to the upper anterior region of her head.
A.
Subdural haemorrhage
B.
Trigeminal neuralgia
C.
Glaucoma
D.
Migraine
E.
Berry aneurysm
F.
Subarachnoid haemorrhage
G.
Tension headache
H.
Sinusitis
I.
Benign intracranial hypertension
Glaucoma
A 60 year old lady presents with a hard palpable mass in the right breast. The mammography shows that the lump is calcified. FNA results show that the cells in the lump have pleomorphic nuclei and that the lump has a necrotic centre
A.
Invasive Carcinoma
B.
Gyaecomastia
C.
Lobular Carcinoma in situ
D.
Pagets Disease of the nipple
E.
Duct Papilloma
F.
Nipple adenoma
G.
Lymphoma
H.
Fibroadenoma
I.
Ductal Carcinoma in situ
Ductal Carcinoma in situ
A 23 year old lady comes to the breast clinic because she is worried of having breast cancer as her mother and grandmother both have had the disease. The mammogram finds no abnormality but you think you feel a number of multifocal lumps
A.
Invasive Carcinoma
B.
Gyaecomastia
C.
Lobular Carcinoma in situ
D.
Pagets Disease of the nipple
E.
Duct Papilloma
F.
Nipple adenoma
G.
Lymphoma
H.
Fibroadenoma
I.
Ductal Carcinoma in situ
Lobular Carcinoma in situ
A 45 year old lady is referred to the breast clinic with nipple retraction
A.
Invasive Carcinoma
B.
Gyaecomastia
C.
Lobular Carcinoma in situ
D.
Pagets Disease of the nipple
E.
Duct Papilloma
F.
Nipple adenoma
G.
Lymphoma
H.
Fibroadenoma
I.
Ductal Carcinoma in situ
Invasive Carcinoma
A 30 year old woman is referred to the breast clinic with a red, roughened and ulcerated nipple.
A.
Invasive Carcinoma
B.
Gyaecomastia
C.
Lobular Carcinoma in situ
D.
Pagets Disease of the nipple
E.
Duct Papilloma
F.
Nipple adenoma
G.
Lymphoma
H.
Fibroadenoma
I.
Ductal Carcinoma in situ
Pagets Disease of the nipple
A 56-year old female presents with a palpable mass in her left breast. Radiology shows microcalcification.
A.
Fine needle aspirate
B.
BRCA1 gene
C.
Hormone replacement therapy
D.
Paget’s disease of the nipple
E.
Carcinoma-in-situ (lobular)
F.
Nulliparity
G.
Axillary lymph node metastases
H.
Fibroadenoma
I.
Carcinoma-in-situ (ductal)
J.
Invasive breast carcinoma
K.
Oestrogen-receptor positivity
Fibroadenoma
involves focal calcification of the
media of small medium-sized arteries. It usually presents in patients
over 50 years of age and unlike atherosclerosis, there is no associated
inflammation in the pathogenesis.
Monckeberg arteriosclerosis
What differentiates dermatomyositis from MG
Sparing of the ocular muscles
45-year-old man is referred to the gastroenterology outpatient clinic due to
severe epigastric pain and an episode of haematemesis. Further testing reveals
he is Helicobacter pylori positive and has a 20 pack–year history of smoking.
Peptic ulcer disease (I) can either be duodenal or gastric. Ulcers differ from
erosions as they extend to the submucosa (sometimes to the muscularis
mucosa) and take weeks to heal, whereas erosions breach the mucosa only
and take days to heal. The main causes of peptic ulcers are Helicobacter
pylori, NSAIDs, Zollinger–Ellison syndrome and smoking. These factors
disrupt the balance between protective (mucus layer and bicarbonate secretion)
and damaging (acid and enzymes) elements leading to ulceration.
A 56-year-old woman is investigated by the hepatology team for decompensated
liver disease. A liver biopsy sample stains blue with Perl’s Prussian blue stain.
A Cholangiocarcinoma
B Cirrhosis
C α1-Antitrypsin deficiency
D Haemosiderosis
E Primary biliary cirrhosis
F Haemochromatosis
G Hepatocellular carcinoma
H Primary sclerosing cholangitis
I Wilson’s disease
Haemochromatosis (F) is an autosomal recessive condition that is due to
a mutation in the HFE gene. The HFE protein regulates iron absorption
that is stored as haemosiderin. Histological features of haemochromatosis
include a golden-brown haemosiderin deposition in the parenchyma
of many organs. Haemosiderin eventually leads to inflammation and subsequent fibrosis. Histological samples of affected tissue will stain blue
with Perl’s Prussian blue. Organs affected include the liver (cirrhosis),
pancreas (diabetes), skin (bronzed pigmentation), heart (cardiomyopathy)
and gonads (atrophy and impotence).
With what is PBC strongly associated?
Sjogren’s
Histological features include periductal fibrosis that eventually
invades the lumen causing concentric onion-ring fibrosis.
PSC
A 54-year-old man is referred to the dermatologist with a brown warty lesion
on his nose which has a rough consistency. Biopsy of the lesion reveals solar
elastosis.
A Pemphigoid
B Bowen’s disease
C Pityriasis rosea
D Lichen planus
E Actinic keratosis
F Psoriasis
G Basal cell carcinoma
H Erythema multiforme
I Malignant melanoma
J Pemphigus
Actinic keratosis
begins as a single scaly macule that is salmon-pink,
<5 cm and known as a ‘herald patch’. After several days multiple small
pink rashes appear posteriorly creating a fir-tree pattern.
Pityriasis rosea
involves the following: haematuria, red cell and
white cell casts, dysmorphic red cells, oliguria and hypertension.
Nephritic syndrome
form as a result of Tamm–Horsefall secretions
in the distal collecting duct and collecting duct that ‘glue’ cells
together,
Cellular casts
it mimics breast cancer; the presentation
may be nipple retraction due to fibrosis and bloody discharge secondary
to rupture of the ducts.
Duct ectasia
usually
occurs mainly in pre-menopausal women and is bilateral and multifocal.
No calcification occurs and hence there is no detection on mammogram.
Histologically there is no necrosis and uniform nuclei are present.
Lobular carcinoma in situ
occur in pre- or post-menopausal
women. They are usually unilateral and unifocal. On mammogram,
microcalcification may be visible secondary to central necrosis.
DCIS
Histological investigation reveals an ‘artichoke-like’
appearance as the stroma pushes up on the epithelium to form clubs.
Phylloides tumour
has a scirrhous look
whereby the centre is very fibrous giving a dense white appearance.
has the worst prognosis compared to all other invasive carcinomas (medullary,
mucinous, tubular and papillary carcinoma).
Infiltrating ductal carcinoma
A 35-year-old man with pain and difficulty bending his left knee. X-ray reveals
many lytic lesions in the epiphysis of the patient’s left knee.
A Osteoporosis
B Fibrous dysplasia
C Paget’s disease
D Osteomalacia
E Osteochondroma
F Osteoid osteoma
G Renal osteodytrophy
H Enchondroma
I Giant cell tumour
Giant cell tumour (GCT; I) is a borderline malignant tumour of giant osteoclast
cells. The cells are similar to those found in Paget’s disease as they
have multiple nuclei (>20). The osteoclastic cells cause lytic lesions in the
epiphyses (especially around the knee) that are visible on X-ray and may
give a characteristic ‘soap bubble’ appearance. Histological features include
multinucleated giant osteoclasts with surrounding ovoid and spindle cells.
Histological features include
multinucleated giant osteoclasts with surrounding ovoid and spindle cells.
visible on X-ray and may
give a characteristic ‘soap bubble’ appearance
Giant cell tumour
An 8-year-old boy has been diagnosed with precocious puberty. A routine
examination by the paediatrician reveals café-au-lait spots on the child’s back.
The boy has had numerous fractures of his femur and tibia bilaterally after falls.
A Osteoporosis
B Fibrous dysplasia
C Paget’s disease
D Osteomalacia
E Osteochondroma
F Osteoid osteoma
G Renal osteodytrophy
H Enchondroma
I Giant cell tumour
Fibrous dysplasia (B) occurs due to the developmental arrest of normal
bone structures secondary to an osteoblast maturation defect. The most
common sites affected are the proximal femur and ribs. On X-ray,
fibrous dysplasia may cause a ground-glass or soap bubble appearance.
Histological investigation reveals trabeculae that lack osteoblastic
rimming. Two possible syndromes can arise: 1) Mono-ostotic (70
per cent) affecting femurs more than ribs occurring in patients under
30 years of age, and 2) McCune–Albright syndrome (30 per cent) that
is poly-ostotic and causes café-au-lait spots and precocious puberty.
A 57-year-old overweight patient suffers an acute myocardial infarction and
subsequently dies. A post-morterm examination of the infarcted area shows
extensive cell infiltration including polymorphs and macrophages. There is also
extensive debris post necrosis and the cytoplasm is homogeneous making it
difficult to see the outlines of the myocardial fibres. There is no evidence of
collagenization or a scar. How long after the initial attack did the patient die?
A At the time of the attack (0–6 hours)
B Hours after the attack (6–24 hours)
C Days after the attack (1–4 days)
D Within the first 2 weeks of the attack (4–14 days)
E Weeks and months after (14 days +)
C In the first 6 hours (A) in the evolution of an acute myocardial infarction
(MI), the histology is normal. Necrotic cell death takes place between
6 and 24 hours (B). Pathologically, there is contraction band necrosis
(dark red/pink wavy lines extending across the myocardial fibres), loss of
nuclei in the myocardial cells and the start of a homogeneous appearing
cytoplasm. At 1–4 days (C) following an acute MI, the start of an extensive
acute inflammatory response takes place with cell infiltration. Debris
is left by the necrosis in the previous stage and the cytoplasm is homogeneous
so that it is difficult to see the outlines of the myocardial fibres.
Infiltration of polymorphs, and later macrophages, takes place. Removal
of the debris takes place at about 5–10 days. At approximately 2 weeks
(D), the area undergoes repair and a classic young scar with new capillaries
is seen with early collagenization. Macrophages and myofibroblasts
are present in large numbers. Some granulation tissue and collagen synthesis
will continue on to the next stage. In the months (E) following an
acute MI, the area starts to strengthen with the formation of a decellularizing
scar. This established scar will be seen as a pale white collagenized
area within the interstitium between myocardial fibres.
A 41-year-old man presents with severe central chest pain which he describes as
‘tearing’ in nature and radiating to the back. He is tall, with long limbs and long
thin fingers. He also has an aortic regurgitation murmur. Histologically there is
cystic medial necrosis in the aortic wall. In which syndrome are these findings
most likely?
A Ortner’s syndrome
B Ehlers–Danlos syndrome
C Down syndrome
D Turner syndrome
E Marfan syndrome
Cystic medial necrosis is a disorder particularly affecting the aorta,
causing focal degeneration of the elastic tissue and muscle fibres in the
media, with accumulation of basophilic ground substance. This leads
to cyst-like pools between the fibres disrupting the normally parallel
arrays. Clinically, aneurysm formation becomes more likely. It is
more frequent after 40 years of age and is twice as common in males.
There is evidence that links cystic medial necrosis to aortic dissection
in patients with a variety of syndromes, the most common of which is
Marfan’s syndrome (E). These patients are characteristically tall with
long limbs and long thin fingers. All other syndromes are inconsistent
with this patient. The most common murmur in Ortner’s syndrome (A)
is mitral stenosis, associated with an enlarged left atrium and recurrent
laryngeal nerve palsy. Turner’s syndrome (D) is only present in females.
Congenital heart defects include: aortic coarctation, aortic stenosis,
ventricular septal defect and atrial septal defect, but aortic dissection is
uncommon. In Ehlers–Danlos syndrome (B), cystic medial necrosis can
cause fragile blood vessels but aortic regurgitation is not commonly
present. Down syndrome (C) patients may have congenital atrioventricular
septal canal defects. Other causes of cystic medial necrosis in patients
without Marfan’s syndrome are advanced age and chronic hypertension.
The activity of the plaques in a 25-year-old multiple sclerosis patient is described
with the presence of oedema and macrophages, and some myelin breakdown.
Which ICDNS (International Classification of Diseases of the Nervous System)
plaque type classification best fits the description?
A Acute plaque
B Early chronic active plaque
C Late chronic active plaque
D Chronic inactive plaque
E Shadow plaque
Multiple sclerosis (MS) is the leading cause of disability in Western
countries among young individuals between 20 and 40 years of age.
The pathological hallmark of MS is the presence of demyelinating
plaques on MRI scanning of the central nerves (spinal cord and brain).
Particular things to look for are myelin loss, destruction of oligodendrocytes,
and reactive astrogliosis, often with relative sparing of the axon
cylinder until later stages. The number of plaques, which range from a
few to several hundreds, is indicative of the severity of the disease.
According to the activity, the International Classification of Diseases
classifies plaques as follows;
Acute plaque (A): Minor changes (e.g. oedema) and often difficult to
recognize
Early chronic active plaque (B): Oedema and macrophages, indicative of
an inflammatory disorder of the central nervous system, with some myelin
breakdown. Reactive astrocytosis is present
Late chronic active plaque (C): Complete loss of myelin. Some macrophages
will contain myelin debris and there will be often very mild perivascular
inflammation at this stage with enlarged perivascular spaces
Chronic inactive plaque (D): Complete loss of myelin with the absence of
macrophages
Shadow plaque (E): Nearly complete remyelination as a thin myelin with some
scattered macrophages and a mild microglial up-regulation.
s a degenerative disease with
cell death of the dopamine-producing neurons of the substantia nigra,
resulting in depigmentation. Furthermore, patients with this condition
are alpha-synuclein and ubiquitin positive.
Parkinson’s disease
characteristically
shows cerebral atrophy in the caudate nucleus and putamen and
several changes in neurotransmitters.
Huntingtons
is associated
with glial cytoplasmic inclusion bodies (i.e. Papp–Lantos bodies).
MSA
is associated with
pernicious anaemia in the elderly and typically affects the body of the
stomach.
Autoimmune chronic gastritis
the dominant feature is the epithelial
change with minimal inflammation that can be idiopathic or due to reflux of bile-containing duodenal fluid or drugs
Reactive gastritis
is the most common form of
chronic gastritis, accounting for 90 per cent of cases, and it is known
that the pyloric antrum is the most severely affected area.
H pylori associated gastritis
is a rare disease characterized by gross
hyperplasia of gastric pits and a marked increase in mucosal thickness
and typically affects the fundus and body of the stomach.
Menetrier’s disease
Ninety to ninety-five per cent
of patients are HLA DR3 and HLA DR4 positive.
T1DM
A 39-year-old man is diagnosed with a colon cancer proximal to the splenic
flexure that is poorly differentiated and highly aggressive. There are no associated
adenomata. It is an autosomal dominant condition that involves gene mutations
of DNA mismatch repair genes. What is the most likely diagnosis?
A Familial adenomatous polyposis
B Gardner’s syndrome
C Colorectal carcinoma
D Hereditary non-polyposis colorectal cancer
E Hamartomatous polyps
18 D Hereditary non-polyposis colorectal cancer (HNPCC) (D) is an uncommon
autosomal dominant disease but the cancers are poorly differentiated and
highly aggressive, therefore screening for identification of carriers for surveillance
is necessary. Familial adenomatous polyposis (FAP) (A) is also
a rare autosomal dominant condition that is caused by a mutation in the
FAP gene on chromosome 5. It is characterized by the presence of adenomata
in the large bowel, which this patient did not have, yet also carries a
90 per cent risk of developing into carcinoma by the age of 45. Gardner’s
syndrome (B) is similar clinically, pathologically and aetiologically to FAP
and also carries a high carcinoma risk. However, there are very distinct
extra-intestinal manifestations of Gardner’s syndrome, including multiple
osteomas of the skull and mandible, epidermoid cysts and desmoid
tumours. Colorectal carcinoma (C) of the sporadic type is inconsistent
with the history and age of the patient, as it is more commonly present
in patients over the age of 60. Hamartomatous polyps (E) often present
in childhood or adolescence as part of Peutz–Jeghers syndrome and these
carry a small risk of carcinomas. Patients also tend to have pigmented
lesions around the mouth that are characteristic of this condition.
A 32-year-old woman presents with generalized fatigue. Full blood count shows
a reduced haemoglobin level and reduced mean corpuscular volume. A peripheral
blood film has revealed iron deficiency anaemia. What features are most likely to
be seen on her peripheral blood film?
A Hypochromic and microcytic red blood cells with anisopoikilocytosis and
acanthocytes
B Hypochromic and microcytic red blood cells with hypersegmented
neutrophils
C Hypochromic and microcytic red blood cells with anisopoikilocytosis and
no evidence of basophilic stippling
D Hypochromic and microcytic red blood cells with Howell–Jolly bodies and
basophilic stippling
E Hypochromic and macrocytic red blood cells with target cells, acanthocytes
and Howell–Jolly bodies
C Features of iron deficiency anaemia are hypochromic (pale) and
microcytic (small) red blood cells. Poikilocytes are red blood cells
that are abnormally shaped. When there are variations in shape and
size, it is known as anisopoikilocytosis. Basophilic stippling (aggregation
of ribosomal
material) is absent in iron deficiency and present in
b-
thalassaemia trait and lead poisoning.
In megaloblastic anaemia, there is impaired DNA synthesis and this can
be caused by B12 deficiency, folate deficiency and drugs. Here the features
are the characteristic hypersegmented neutrophils and macrocytic
red blood cells.
In hyposplenism, there is presence of target cells known as codocytes
(red blood cells that have a high surface area:volume ratio).
Acanthocytes (spiculated blood cells/spur cells) and Howell–Jolly bodies
(nuclear remnants visible in red cells) are also present in the hyposplenism
picture.
A 27-year-old woman has developed pain in her right proximal femur. She has
a history of intermittent hip pain since childhood. An X-ray has demonstrated a
‘soap bubble’ appearance indicative of osteolysis and a characteristic shepherd’s
crook deformity. The biopsy would show irregular trabeculae of woven bone said
to resemble Chinese letters. What is the most likely diagnosis?
A Non-ossifying fibroma
B Fibrous dysplasia
C Giant cell reparative granuloma
D Ossifying fibroma
E Simple bone cyst
24 B Fibrous dysplasia (B) is a benign disorder of children and young adults,
whereby lesions composed of fibrous and bony tissue develop usually
in the ribs, femur, tibia or skull. It mostly presents as bone pain
and weakness in female patients under 30 years of age and results
from congenital dysplasia of bone, consistent with the patient’s history.
There are three forms: the more common monostotic form
(lesions localized to only one bone), polyostotic (multiple lesions) and
McCune–Albright syndrome, which also has endocrine manifestations.
Shepherd’s crook deformity refers to a varus angulation of the proximal
femur commonly seen in the femoral involvement of polyostotic
fibrous dysplasia. Histologically, it is characterized by loose fibrous tissue
with metaplastic immature or woven bone trabeculae arranged in a
‘Chinese letters’ formation.
Non-ossifying fibroma (A) is often asymptomatic and merely an incidental
finding on X-ray and occurs in an even younger group of
patients. Ossifying fibroma (D) is a benign, fibrous tumour with reactive
bone formation that shows local aggressive behaviour. Lesions are
found in the mandible for adults, and the tibia for children. Giant-cell
reparative granuloma (C) is an uncommon benign reactive intraosseous
lesion and can occur in the skull, jaw, hand and foot. The histology
resembles giant cell tumour of bone (see below). A simple bone cyst (E)
is common in the proximal metaphysis of the humerus and asymptomatic
unless there has been a fracture.
A 36-year-old man presents with swelling of his middle finger and subsequently
a fracture. His X-ray shows cotton wool calcification and histopathology shows
evidence of a tumour composed of benign hyaline cartilage. It is believed that he
has only a very slight risk of malignant transformation. What is the most likely
diagnosis?
A Osteochondroma
B Multiple myeloma
C Osteoid osteoma
D Giant cell tumour
E Enchondroma
25 E Enchondroma (E) is a benign intramedullary cartilage tumour usually
found in the central mature hyaline cartilage of the short tubular bones
of the hands and feet. It may present at any age (average is 40 years)
and is often asymptomatic, but some patients present with pain, fracture
or swelling of the affected area. There are lytic lesions on X-rays that
usually contain variably calcified chondriod matrix and histopathological
findings showing bluish-grey lobules of hyaline cartilege. There may
be a thin layer of lamellar bone surrounding the cartilage nodules but
no permeation of pre-existing host bone which is a positive sign that the
lesion is benign. Each potential enchondroma needs to be evaluated on
imaging and histology to distinguish it from low-grade chondrosarcoma.
Osteochondroma (A) is the most common skeletal neoplasm, often affecting
boys in their teens and often found in the long bones. When the
growth plates close in late adolescence, there is usually no further growth
of the osteochondroma. There is a low risk of malignancy in osteochondromas
associated with syndromes that involve multiple lesions such as
Ollier’s disease or Maffucci’s syndrome. Osteoid osteoma (C) is a small,
benign lesion surrounded by a zone of reactive bone. It mostly occurs in
the long bones, with a very distinct clinical picture in that pain is dull,
worse at night and relieved by aspirin. Similar radiology and histology
are seen in osteoblastomatomours that are larger than 1 cm diameter and
that commonly affect the vertebrae. Pathological fracture is a common
presentation of giant cell tumour (D), which is more common in females,
is locally aggressive and can metastasize. It does not occur in the immature
skeleton. It is more common in the metaphysics of the long bones,
especially the distal femur and proximal tibia. There is no calcification
on X-ray and, histologically, there would be numerous multinucleated
giant cells. The mono-nuclear cell population is the neoplastic cells.
Multiple myeloma (B) is highly unlikely as it is a different classification
of bone tumours altogether and is considered as a blood cell malignancy
rather than a primary bone tumour. It is a malignant tumour of plasma
cells that causes widespread osteolytic bone damage. It may be in are
bone only when it is known as plasmacytoma rather than myeloma.
