Hemoglobin Variants Flashcards
most Hb has what subunits?
a2B2
Hb can be composed of what polypeptides?
alpha, beta, gamma, delta
what chromosomes and gene clusters produce the polypeptides (chain) assembled into various hemoblogins?
- chromosomes 11 and 16
- chromosome 11
- alpha like cluster –>
- alpha chains
- zeta chains
- alpha like cluster –>
- chromosome 16
- beta like gene cluster –>
- beta chains
- gamma chains
- delta chains
- epsilon chains
- beta like gene cluster –>
- chromosome 11

discuss the Hb variants present throughout development, and what
- poypeptides they consist of
- genes those polypeptides came from
- chromsomes those genes are on
- embryonic = alpha, gamma, zeta and episilon polypeptides used.
- zeta (from alpha cluster) and episilon (from beta cluster) are ONLY seen in embryonic.
- combos:
- zE (gower 1)
- aE (gower 2)
- portland (zy)
- fetal = a2y2
- adult = a2B2 > a2d2
the globin genes are activated in what direction along the chromosome? what effect does this have?
5’ –> 3’ direction.
- polypeptides close to the 5’ end seen in earlier in life (zeta, episilon, gamma) and the 3’ end seen later in adulthood (beta, delta)

what leads to hereditary persistence of fetal Hb? what are the clinical imlications of this condition?
- usually due to improper expression/deletion of the beta and delta globin genes, which are:
- found closer to the 3’ of their chromosomes & normally expressed only in adulthood.
- as a result, embryonic/ fetal chains (z, E, y) persist in the blood.
- sustained levels of fetal Hb can be useful:
- sickle cell
- B-thalassemia
- sustained levels of fetal Hb can be useful:
- as a result, embryonic/ fetal chains (z, E, y) persist in the blood.
- found closer to the 3’ of their chromosomes & normally expressed only in adulthood.
discuss the solubility of Hb & its significance
- the Hb tetramer is highly soluble
- however, individual subunits are not.
- so, is subunits are not expressed in sufficient amounts/improperly assembled they can precipiate –> inclusions
discuss the primary, secondary, tertiary & quaternary strucutres of globins
- primary: aa sequence - highly similar for each globin (polypeptide)
- secondary: rich in alpha helices
- tertiarty: hyrophobic aas in core, hydrophillic aas out
- quaternary: two aB dimers (or other variation)
Hb quaternary structure is highly dependent on what interactions?
- proper hydrophobic/hydrophillic aa alignment
- a1b1 & a1B2 contant points
- His residue in F helix
how many amino acids are there?
20
what type of mutation causes sickle cell anemia (HbS)?
point mutation:
-
Glu –> Val in B subunit
- this is a loss ot two negative charges:
- Glu = -, hydrophillic
- Val = neutral, hydrophobic
- this is a loss ot two negative charges:
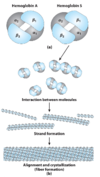
discuss the pathogenesis of sickle cell anemia
- Glu –> Val mutation.
- This Val residue is on the surface of the T-state (tense) molecule
- Val residue interacts with other hydrophobic aas (phenylalinine, leucine) –> Hbs molecules aggregate
- 14 HbS total HbS fiber
why is HbS aggregates less likely to form when HbS is oxygenated?
- when oxygenated form, HbS is the in R-state.
- the arrangement of aas shifts such that key hydrophobic residues (F, L) available to interact with mutant Val in the T-state become buried in the HbS core.
- Val cannot make the non-polar bonds that lead to aggregation & HbS fiber formation.
how to dx sickle cell anemia?
- by electrophoresis
- due to structural HbS migrates differently than normal adult forms of HbA - a2B2, a2D2
- the abnormal beta subunit (S)
- due to structural HbS migrates differently than normal adult forms of HbA - a2B2, a2D2

heinz bodies
- defintition
- what forms them?
- how are they detected?
- clinical significance?
- inclusions of denatured, aggregated Hb (typically Hb variants) within RBCs
- clinical:
- congenital heinz body hemolytic anemia (CHBA) - these RBCS are more susceptible to hemolysis
- dx - smears stained for reticulocytes