Hematopoiesis - Strom 03.16.15 Flashcards
What are pluripotent stem cells?
- Defined by ability to “salvage” all elements of hematopoiesis in recipient after their bone marrow contents are wiped out by irradiation or chemotherapy
- Rare (1 in 20 million), and express receptors for key growth factors
- Morphologically, “blasts” (small round cells with hypodense chromatin and no morphologic features associated w/differentiation), but make up only a tiny fraction of the morphologic blasts in the bone marrow (so they can’t be ID’d clearly by morphology)
- In research settings and gene therapy protocols they can be purified almost to homogeneity by identifying certain characteristic cell surface markers
Can you use a microscope to pick out burst (BFU) and colony forming units (CFU) from the bone marrow? How are they functionally defined?
- NO - although it would be really helpful if you could
- Functionally defined by the fact that if you dump a mixture of cells from bone marrow into petri dish, a particular cocktail of growth factors causes formation of colonies with morphologic and immunophenotypic features of maturing cells in specific lineages
What are the 4 key heme growth factors?
- Thrombopoietin (TPO)
- Erythropoietin (EPO): this drug is the reason many pts with renal failure are alive now
- Granulocyte-macrophage colony stimulating factor (GM-CSF)
- Granulocyte colony stimulating factor (G-CSF)
What percentage of cells in the bone marrow are blasts? What is the breakdown within this group of cells?
- Blasts <4%
1. Erythropoiesis: 20-30% (incl. thrombopoiesis)
2. Myelopoiesis: 60-70%
3. Lymphopoiesis: 10-20%
4. Pluripotent: probably <0.1% - NOTE: microscopic view doesn’t quite match this b/c can’t identify the CFU/BFU cells morphologically; most likely have no features associated w/differentiation at all – that is, they are morphologic “blasts”
Describe the normal morphologic maturation of granulocyte precursors.
- Continuous process, but represented categorically:
Blast (3-4%) -> Promyelocyte (2-8%) -> Myelocyte (10-13%) -> Metamyelocyte (10-15%) -> Bands and neutrophils (25-40%)
- More mature species outnumber less mature species b/c granulocyte maturation a process of differentiation and cell division; cells thought to lose ability to divide only after the myelocyte stage
- Can be affected by # of disease states, e.g., it can INC in overall numbers, be shifted to the left or right, or be blocked part way through (“maturation arrest”)
What are the key regulators of granulopoiesis?
- GM-CSF: how its production is tied in to the multiple biological factors that can stimulate increase in neutrophil count not well understood
- Acts well up the differentiation chain, such that it will also stimulate production of eosinophils (which branch off this process b/t morphologic blasts & promyelocytes)
- G-CSF: acts more specifically on neutrophil precursors -> exact morphologic stage of the least differentiated cell it acts on is not clear
- NOTE: both of these factors released via bone marrow stromal cells; aka, myelopoiesis
Describe the normal maturation of red cell precursors.
- Epo production stimulated by hypoxia, sensed by renal peritubular cells -> renal failure usually results in anemia (add to differential diagnosis of unexplained anemia)
- Blast (3-4%) -> Pronomoblast -> Basophilic erythroblast -> Polychromatophilic erythroblast -> Normochromic erythroblast
- 4 or 5 cell divisions, but red cell precursors seen in bone marrow mostly most mature erythroid precursors, “normochromic erythroblasts”
1. After 5th division, nuclei extruded and nascent red cells released (polychromasia, reticulocytes)
Describe thrombopoiesis (megakaryocytes).
- Platelet production very different than RBC, leukocyte production -> megakaryocytes: polyploid factory cells (nuclei divide multiple times, so instead of normal diploid (2N) state, contain 16-32 haploid genomes)
- Megakaryocytes extend snakelike tubes called “proplatelets” into highly perforated (fenestrated) blood vessels in bone marrow (sinuses) -> mature platelets lopped off one at a time from ends of the proplatelets
- Regulated by hepatocytes and other sources (blast, 3-4% -> immature -> mature)
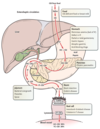
Where is TPO made? What does it do?
- Thought to be made at a constant rate, primariily in liver
- Binds both platelets and megakaryocytes; when bound to megakaryocytes, stimulates their production from immature precursors and platelet production from mature megakaryocytes
- Low platelet count allows more TPO to bind megakaryocytes, stimulating more thrombopoiesis
What is the basic structure of the EPO and TPO receptors? What would you expect if both of these were stuck in the ON position?
- EPO (expressed on erythroid precursors) and TPO (aka, c-MPL) receptors (on mature/immature megakaryocytes) have very similar structures and functions
- Both transmembrane dimers with two copies of relatively inactive kinase (Jak-2) attached to their cytoplasmic tails
- If stuck in ON position, you would expect: BLOOD CLOTS
What happens when the Type 1 hematopoietic growth factor receptors are activated? Why is it important to understand this?
- Cytokine binding to receptor swings cytoplasmic tails, and their bound kinases (Jak-2), close together -> kinases phosphorylate each other, increasing activity
- More active kinases phosphorylate other targets further down cytoplasmic tail, initiating series of other signal transduction steps (& altered transcription patterns) w/divergent actions in different lineages (EPO vs. TPO)
- Both processes result in differentiation, proliferation of precursors in their respective lineages
- Acquired mutations in receptors, or Jak-2 kinase, are clinically relevant – paticularly if they cause constitutive activation of these signal transduction pathways
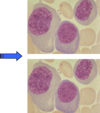
What happens to bone marrow cellularity with age?
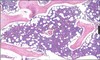
It declines. The image shown is a bone marrow biopsy from a 5 y/o.
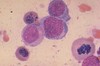
What is this?

High power view of bone marrow biopsy. You can see some erythroid (top right), myeloid (middle), and megakaryocyte (middle) cells here.
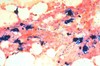
What is this? Is it normal or abnormal? Why?
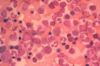
- Aspirated (sucked out with needle) normal bone marrow material
- Should be able to identify the neutrophils and bands present, and the smaller cells with dark and very round nuclei (mostly normochromic erythroblasts)
- More important point is that there are a lot of different cell morphologies present, making this NORMAL -> when you see bone marrow aspirate containing predominantly one cell type, there’s a problem
What is heme? What does heme synthesis require?
- Heme is an oxygen carrier
- Requires iron, B6, succinyl CoA, and glycine (which requires B12 and folate)
What does globin production require?
- Requires amino acids of course, and malnutrition definitely causes anemia
- In this country, problems with globin production more commonly due to problems with the genes for its two component proteins (alpha and beta), also required for synthesis
What is required for red cell DNA synthesis?
- Requires deoxynucleoside triphosphates, which require ribonucleotide reductase and thymidine (B12 and folate)
- Although thrown away at end of process, nucleus is replicated multiple times in red cell production, so the production process kind of counterintuitive – red cells lack nuclei, but you have to make tons of DNA in order to produce red cells (again requiring both adequate nutrition and a couple of key cofactors (vitamins))
How is the red cell production process regulated?
- Primarily by erythropoietin
- Will fail if kidneys fail, or if bone marrow gets filled up by cells that don’t belong there (which can include fibroblasts and the collagen they produce)
Is anemia a diagnosis? Why or why not?
- Anemia might have been a diagnosis once, but it is not now - any more than fatigue or pallor or tachycardia are diagnoses
- Anemia is a laboratory finding; it remains undiagnosed until you find a cause for it
What are the 3 possible disease mechanisms for anemia?
- Losing red cells from the bloodstream
- Not making enough red cells
- Both
What is the morphology of iron deficiency?
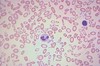
- Morphology: small red cells without much hemoglobin, i.e., microcytosis and hypochromia; lots of variation in red cell size & shape, i.e., anisocytosis & poikilocytosis
1. Hypochromia will be seen a bit earlier than anisocytosis and poikilocytosis, i.e., when pt’s anemia only mild or moderate - Area of central pallor in biconcave discs is enlarged; rule of thumb is that if central pallor diameter > 1/3rd of red cell diameter, cell is hypochromic, or lacking color - i.e. lacking hemoglobin
- Anemia, microcytosis, hypochromia suggest iron def, but other problems can yield a similar picture -> to confirm iron deficiency, you’ll need additional lab tests
What are anisocytosis and poikilocytosis? When will these terms show up in your pt’s lab results?
- Anisocytosis: variation in red cell size
- Poikilocytosis: variation in shape
- Descriptive terms that will show up in lab results IF a manual differential count is done; an automated CBC will not generate these terms, but hematology analyzer will tell you if red cells size distribution is increased (“red cell distribution width,” or RDW), which correlates well with morphologic anisocytosis.
- Both are characteristic of (but not specific for) severe iron deficiency
Describe the process by which dietary iron gets into circulation from the gut (receptors covered in other card).
- Dietary iron mostly in oxidized (ferric) state; reduced (in duodenum) before being taken up (in rest of sm bowel)
- Ascorbate (vit C) reducing agent duodenal reductase (duodenal cytochrome b) uses (to reduce one thing you have to oxidize another) -> uptaken iron re-oxidized for transport (via serum oxidase, ceruloplasmin)
- Free iron in plasma would be bad thing, augmenting bacterial growth and catalyzing formation of superoxide radicals from oxygen -> so, transport handled by specialized circulating protein, transferrin, which only works with Fe+++ (it can’t transport ferrous iron)
What transporters are involved in iron absorption by gut enterocytes?
- Multistep process with 2 transporters: 1 on luminal mem (DMT-1) and 1 on basement mem (ferroportin)
- Both transporters regulated by physiologic conditions
1. DMT-1: iron-dependent regulation of its mRNA translation and stability
2. Ferroportin: regulatory peptide hepcidin; also responsible for exporting iron from macros that hold body’s iron storage pool– and hepcidin regulates it in that location as well