Hematopathology - Anemia & Hemostasis Flashcards
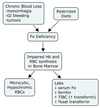
This anemia typically results from chronic blood loss via GI ulcers or bleeding, mehorrhagia, and some tumors.
Serum Fe and ferritin levels are decreased, while TIBC is increased because of increased serum transferrin. %saturation of transferrin is low.
It is characterized by microcytic, hypochromic anemia.
In a severe form, it can produce atrophic glossitis.

Fe-deficiency anemia
Because of the production defect, there is no reticulocytosis.
This anemia arises in association with chronic inflammatory diseases or malignant conditions, which cause an ineffective use of iron from macrophage stores in bone marrow.
Therefore, Fe stores are normal or elevated, as shown by Prussian blue staining.
Serum Fe and TIBC are both decreased.
Anemia of Chronic Disease
(tuberculosis, cancers, rheumatoid arthritis…)
*Anemia of Chronic Disease
–Serum Fe and TIBC are both LOW
*Iron-Deficiency Anemia
–Serum Fe is LOW, but TIBC is HIGH
This disorder features hypocellular bone marrow and pancytopenia.
It is sometimes a result of toxic injury, but more often is idiopathic.
Despite elevated EPO levels, reticulocytosis is not present. This underscores the stem cell defect.
Patients present with weakness, fatigue, infection, and bleeding.

Aplastic Anemia
This anemia involves selective suppression of committed erythroid precursors in the bone marrow.
Overall BM cellularity is normal, but there is an absence of erythroid precursors.
Despite elevated EPO, reticulocytosis is absent.
Sometimes it is caused by parvovirus B19.
Pure Red Cell Aplasia
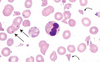
This anemia is caused by impaired DNA synthesis, due to deficiency of vitB12 or folic acid.
Defective DNA synthesis leads to nuclear-to-cytoplasmic asynchrony – leading to formation of large, nucleated erythrocyte precursors.
There is ineffective hematopoiesis: the BM displays hematopoietic activity, but most precursors are not mature and thus undergo intramedullary cell death.
Serum LDH levels are elevated.

Megaloblastic Anemia
[circulating neutrophils often show hypersegmentation]

This form of megaloblastic anemia is an autoimmune disorder in which patients have antibodies against parietal cells and intrinsic factor.
[image: hypercellular BM with megaloblastic erythroid precursors]

Pernicious Anemia
[image: hypersegmented granulocytes]

In a patient with megaloblastic anemia, how can you determine the etiology (vitamin B12 or folic acid deficiency) based on onset and clinical symptoms?
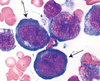
Megaloblastic Anemia
Folate Deficiency
-faster onset (months)
Vitamin B12 Deficiency
- slower onset (years)
- neurologic symptoms due to demyelination of spinal cord columns
In this condition, blood smears show hypochromic and microcytic RBCs with anisocytosis (uneven size and shape), poikilocytosis (abnormal shape), and target cells.
There is increased fetal Hb, which, in addition to the anemia, caues impaired O2 delivery.
BM erythroid hyperplasia ensues, resulting in expanded marrow space that produces cranial and facial bone deformities.

Beta-thalassemia
- Caused by point mutations in the beta-globin gene.
- The homozygous form is more severe.
- The heterozygous form confers a protective effect against malaria.

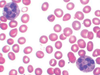
This anemia is a mild microcytic anemia, and its only hematologic abnormality is a small amount of Hb-Bart, which is detectable only in infancy.
Silent-carrier alpha thalassemia
A 10-year-old boy presents with chronic fatigue, splenomegaly, and slight jaundice. Peripheral blood smear shows hyperchromic (no central pallor) spherocytes, along with polychromasia and reticulocytosis. The spherocytes display osmotic fragility. The BM displays erythroid hyperplasia.

Hereditary Spherocytosis
- deficiency of a cytoskeletal component leads to uncoupling of lipid bilayer from cytoskeleton
- 50% patients develop cholelithiasis
[image reveals frequent spherocytes with decreased diameter and lack of central pallor]

This hemolytic anemia is caused by abnormal polymerization of spectrin molecules. Peripheral blood smear shows elliptical or oval eryhtrocytes that have parallel sides. Central pallor is still present.
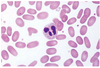
Hereditary Elliptocytosis
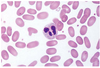
A 45-year-old chronic alcoholic presents with mental confusion. The peripheral blood smear red cells lack central pallor and display irregular spikes on the surface.
The cause of this mild hemolytic anemia is chronic liver disease, in which increased free cholesterol is deposited within cell membranes.

Acanthocytosis
Acanthocytes = “spur cells”

A previously healthy man of Italian origin develops hemolytic anemia after eating a lot of Fava beans. A direct Coombs test is negative.
Fill in the blanks with regards to this man’s G6PD deficiency…
Oxidative stress leads to formation of _____, which cannot transport oxygen, is unstable, and precipitates in the cytoplasm as _____. After passage through the spleen, red cells may have part of their membrane removed, forming _____.
G6PD Deficiency
Oxidative stress leads to formation of methemoglobin, which cannot transport oxygen, is unstable, and precipitates in the cytoplasm as Heinz bodies. After passage through the spleen, red cells may have part of their membrane removed, forming bite cells.
X-linked disorder, so full expression occurs only in males.
The clinical picture of this hemoglobinopathy is dominated by vaso-occlusive disease. Rigid erythrocytes result in obstruction of microcirculation, with subsequent ischemic injury. The rigid erythrocytes are also susceptible to hemolysis.
Infection with Parvovirus B19 can cause an aplastic crisis.
A sequestration crisis occurs when erythrocytes pool in the spleen.

Sickle Cell Anemia
[sickled cells and target cells are evident]
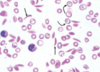
A patient presents with malaise, exudative pharyngitis, and generalized lymphadenopathy. A CBC demonstrates atypical lymphocytes, and the monospot test is positive. He complains of pain and tingling in his finger tips upon exposure to cold temperatures.
The image below demonstrates RBC clumping caused by cold agglutinins.
Cold Antibody Autoimmune Hemolytic Anemia
In immune hemolytic anemias, hemolysis is caused by antibodies against antigens at the erythrocyte surface.
Cold agglutinins may be idiopathic or develop secondary to infections (EBV) or lymphoproliferative disorders.
This hemolytic anemia features spherocytes and polychromasia, but the direct Coombs test is positive. It is usually the result of IgG directed against erythrocyte membrane antigens, such as Rh group antigens.
Warm Antibody Autoimmune Hemolytic Anemia
The direct Coombs test is positive and helps to distinguish immune from nonimmune spherocytosis. In the direct Coombs test, the patient’s RBCs are incubated with antihuman globulin serum. Agglutination (positive result) indicates antibody is present on the cell surface.
This hemolytic anemia reflects incompatibility of blood types between a mother and her developing fetus. Maternal IgG alloantibodies cross the placenta and cause hemolysis of fetal RBCs, resulting in a compensatory release of erythroblasts from fetal bone marrow.
The peripheral blood smear shows nucleated erythroblasts.

Hemolytic Disease of the Newborn

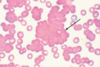
Classic examples of this anemia include DIC and TTP (thrombotic thrombocytopenic purpura).
Intrinsically normal RBCs are fragmented due to mechanical disruption (prosthetic valve, vascular graft) or alteration of endothelial surface (malignant hypertension, vasculitis syndromes). This results in fibrin deposition and platelet aggregation.
The blood smear shows schistocytes and polychromasia.
Microangiopathic Hemolytic Anemia
curved arrows are schistocytes (fragmented RBCs)
straight arrows are Howell-Jody bodies

A child presents with palpable purpura of the lower extremities and buttocks, and arthralgias.
This disease is the most common form of vasculitis in children. It results from immunologic damage to vessel walls, and often follows viral infections.
Histologically, it is characterized by leukocytoclastic vasculitis associated with IgA immune complexes.
Renal disease is a common systemic sign of the disease.
Henoch-Schonlein Purpura
In a child, this disease presents as sudden onset of petechiae or purpura following viral infection. Thrombocytopenia is present, and BM reveals increased megakaryocytes.
In adults, this manifests as bleeding episodes (epistaxis, mehorrhagia, ecchymoses) or excessive bleeding after minor trauma (tooth extraction). It is often associated with collagen vascular diseases (eg, SLE) or CLL.

Idiopathic/Immune Thrombocytopenic Purpura
- reflects Ab-mediated destruction of platelets or their precursors
- autoantibodies are usually IgG, but sometimes IgM
This disease presents with a pentad that includes:
1) fever
2) thrombocytopenia
3) microangiopathic hemolytic anemia (jaundice, schistocytes on blood smear)
4) renal impairment (proteinuria, hematuria)
5) neurologic symptoms (seizures, mental status)
The classic histologic finding is widespread deposition of PAS-positive hyaline microthrombi in arterioles and capillaries of the heart, brain, and kidneys.
PT, PTT, and fibrinogen levels are normal.
[microthrombi]

Thrombotic Thrombocytopenic Purupra
[schistocytes shown in image]
The normal PT, PTT, and fibrinogen levels distinguish this disease from DIC.
ADAMTS13 protein is absent or defective, resulting in uncleaved vWF….leads to platelet aggregation.

This disease results from a genetic defect involving the factor VIII gene.
It presents as spontaneous bleeding.
It is the most common X-linked inherited bleeding disorder.
Hemophilia A
This disease is caused by mutations in the gene encoding for factor IX, a vitamin-K dependent protein that is made in the liver.
It presents as bleeding tendencies.
Hemophilia B
This disease presents as immediate, mucocutaneous bleeding such as easy bruising, epistaxis, GI bleeding, and menorrhagia. Sometimes the presenting symptom is excessive bleeding after surgery.
von Willebrand Disease
This disease typically occurs as a complication of massive trauma, burns, or sepsis.
Patients present with anemia, thrombocytopenia, and widespread purpura.
Other symptoms include:
1) neurologic - seizures, coma
2) renal - mild azotemia to acute renal failure
Findings include elevated fibrin split products, and prolonged PT and PTT.
There are widespread microthrombi.

Disseminated Intravascular Coagulation
Symptoms of DIC reflect microvascular thrombosis and bleeding.

Clinical manifestations of this condition include: arterial and venous thrombosis; spontaneous abortions; and immune-mediated thrombocytopenia or anemia.
Laboratory findings may include:
1) detection of lupus anticoagulants
2) anti-cardiolipid or anti-phospholipid (aPL) antibodies
3) anti-beta2 glycoprotein antibodies
Antiphospholipid Antibody Syndrome
Thrombocytopenia is defined as any platelet count under ?
150,000/µL
Normal levels for erythrocytes


Normal levels for leukocytes


