✅ Headache / Stroke / Meningitis / Seizure / MG Flashcards
Brain death
Brain death is defined as the irreversible cessation of brain activities. Interestingly, the criteria for brain death are not uniform in different countries, but the essential elements in all such criteria include: (1) evaluating cortical and brain stem functions, and (2) proving the irreversibility of brain activity loss (e.g., sufficient observation length, no hypothermia, etc.).
Brain death is a clinical diagnosis. The characteristic findings are absent cortical and brain stem functions. The spinal cord may still be functioning; therefore, deep tendon reflexes may be present. An isoelectric EEG can be used as a confirmatory test, but it is not absolutely necessary. Other diagnostic tools (e.g., Doppler ultrasonography, angiography) can demonstrate cerebral blood flow cessation, but these are not commonly employed.
Myoclonus
Myoclonus is characterized by sudden, involuntary muscle contraction or relaxation that results in movement of limbs or joints. It can occur due to genetic disorders, seizures, medications, or prolonged hypoxia.
- Myoclonus status epilepticus (MSE) is the acute form of PHM that typically develops within 24 hours after the initial hypoxic insult while the patient is still unconscious. It is characterized by a generalized (often symmetric) myoclonus that typically involves the axial, limb, and facial muscles; intermittent eye opening, upward gaze deviation, and swallowing movements are also common. Persistent MSE is considered a marker of poor prognosis.
- Lance-Adams syndrome, the chronic form of PHM, presents days to weeks after the initial insult once the patient has regained consciousness. It is typically focal in nature and exacerbated by action; negative (relaxation) myoclonus also occurs, leading patients to drop objects or fall.
An electroencephalogram should be obtained to help distinguish between other forms of seizure and PHM. The management of both the acute and chronic forms of PHM involves administration of antiepileptic agents (eg, clonazepam, levetiracetam) and supportive care.
HEADACHE
Location:
Unilateral: headache is an invariable feature of cluster headache and occurs in the majority of migraine
attacks
Bilateral: Most patients with tension-type headache report bilateral pain.
Ocular or retroorbital pain: suggests a primary ophthalmic disorder such as acute iritis or glaucoma, optic
(II) nerve disease (eg, optic neuritis), or retroorbital inflammation (eg, Tolosa–Hunt syndrome). It is also
common in migraine or cluster headache.
Paranasal pain localized to one or several sinuses, often associated with tenderness of the overlying periosteum and skin, occurs with acute sinus infection or outlet obstruction.
Focal headache may result from intracranial mass lesions, but even in such cases it is replaced by bioccipital and bifrontal pain when the intracranial pressure becomes elevated.
Bandlike or occipital discomfort is commonly associated with tension-type headache. Occipital localization can also occur with meningeal irritation from infection or hemorrhage and with disorders of the joints, muscles, or ligaments of the upper cervical spine.
Pain within the first (V1) division of the trigeminal nerve, characteristically described as burning in quality,
is a common feature of postherpetic neuralgia.
**Lancinating pain localized to the second (V2) or third (V3**) division of the trigeminal (V) nerve suggests trigeminal neuralgia (tic douloureux).
The pharynx and external auditory meatus are the most frequent sites of pain caused by glossopharyngeal neuralgia.
Red Flags
(Tumor)
Warning signs include:
- Onset after the age of 50 years
- Very sudden onset
- Increase in severity or frequency, with signs of systemic disease.
- Focal neurologic symptoms
- Papilledema
- Headache after trauma
Other “red flags” potentially indicating a secondary headache include: dizziness, lack of coordination, tingling, awakening from sleep due to headache, focal neurologic findings, papilledema, fever, neck stiffness, meningeal signs, tenderness or diminished pulse over the temporal artery, diastolic blood pressure >120 mm Hg, or decreased visual acuity.
Dx: MRI of the brain is preferable to computed tomography of the head in the evaluation of subacute or chronic headache because of improved sensitivity resulting from superior anatomic resolution.
Intracranial neoplasm
Worse on awakening; generally progressive. Headache aggravated by coughing, straining, or changing position.
“Red flag” symptoms: Awakening from sleep due to headache; focal neurologic findings; more general neurologic findings such as dizziness, lack of coordination, or tingling; fever; neck stiffness or meningeal signs; tenderness or diminished pulse over the temporal artery; diastolic blood pressure >120 mm Hg; or papilledema or decreased visual acuity.
Dx: MRI of the brain is preferable to computed tomography of the head in the evaluation of subacute or chronic headache because of improved sensitivity resulting from superior anatomic resolution.
Migraine
Hx:
Migraine With Aura (Classic Migraine): In 30% of migraine patients, headache is preceded by transient neurologic symptoms (aura). The most common auras are visual alterations, particularly hemianopic field defects and scotomas (blind spots) and scintillations (flickerings) that enlarge and spread peripherally. Photopsia (sparks or flashes of light), fortification spectra (arcs of flashing light that often form a zigzag pattern), or a scotoma (blind spots), scintillations (flickerings). Tingling is also possible A throbbing, unilateral (hemicranial) headache ensues.
The duration of episodes is greater than 2 hours and less than 1 day in most patients. During the headache, prominent associated symptoms include nausea, vomiting, photophobia, phonophobia, irritability, osmophobia, and lassitude. Vasomotor and autonomic symptoms are also common. Light-headedness, vertigo, ataxia, or altered consciousness may occur in basilar migraine, which can be distinguished from stroke by both the gradual onset (“migrainous march”) and spontaneous resolution of symptoms. Migraine occasionally produces neurologic deficits that persist into or beyond the pain phase (eg, hemiplegic migraine) and may rarely cause stroke.
Migraine without aura (Common Migraine): is much more common than migraine with aura and produces headache that is most often bilateral and periorbital in location. The pain may be described as throbbing, particularly when severe. Nausea, vomiting, and photophobia are common.
Common dietary triggers include caffeine; nitrates or nitrite preservatives; phenylethylamine, tyramine, and xanthine in aged cheese, red wine, beer, champagne, and chocolate; monosodium glutamate (food additive); dairy products; and fatty foods.
Pain Features: (need 2/4)
POUND: Pulsatile quality (headache described as pounding or throbbing), One-day duration (episode may last 4-72 hours if untreated), Unilateral in location, Nausea or vomiting, and Disabling intensity (altered usual daily activities during headache episode).
Tx: Divided into measures used to abort attacks in progress (abortive) and to prevent future episodes (prophylactic).
Abortive or acute therapy for migraines is appropriate monotherapy if attacks occur less than two to four times per month.
Abortive: Mild attacks are effectively treated with nonsteroidal anti-inflammatory drugs (NSAIDs) or acetaminophen; more severe attacks are treated with a triptan (selective serotonin receptor agonist)[first line] [contraindicated in the presence of ischemic vascular disease and uncontrolled hypertension]. Ergot alkaloids (Dihydroergotamine) is an alternative (contraindicated in CAD and pregnancy).
Approximately 38% of patients with migraine need preventive treatment, but preventive medications are prescribed to only 3% to 13% of these patients.
Prevention (prophylactic): β-blockers (such as propranolol, metoprolol, or timolol), tricyclic antidepressants (such as amitriptyline), and anticonvulsants (such as valproate, topiramate, or gabapentin). Verapamil is the only calcium channel blocker that studies show to have a prophylactic effect [limited]. Amytriptyline therapy begins with a low dose (10 mg at night) and can be titrated up to the most effective dose that does not cause prohibitive side effects (up to 150 mg)
Some herbal products such as feverfew, butterbur root, the mineral magnesium, the vitamin riboflavin, and the antioxidant coenzyme Q10 may have some efficacy in migraine prevention.
Tension-type headache
Episodic tension-type headache is defined as episodes of recurrent nondisabling headache lasting 30 minutes to 7 days.
Hx: Bilateral, pressure/tighteness pain of mild to moderate intensitydiscomfort not aggravated by physical activities and without nausea.
Bandlike or occipital discomfort is commonly associated with tension-type headache. Occipital localization can also occur with meningeal irritation from infection or hemorrhage and with disorders of the joints, muscles, or ligaments of the upper cervical spine.
Tx:
Drug treatment usually begins with NSAIDs. Aspirin and acetaminophen are more effective than placebo in treating these headaches, but comparative studies suggest superior benefit from the nonsteroidal anti-inflammatory drugs (NSAIDs) ibuprofen and naproxen.
The addition of caffeine to aspirin or NSAIDs increases treatment efficacy. Prophylaxis, often with a tricyclic antidepressant, may be needed.
Tricyclic antidepressants, such as amitriptyline, are commonly used for prophylaxis for tension-type headache
Trigeminal autonomic cephalalgias:
Rare. Group of primary headache disorders characterized by excruciating unilateral headache that occurs in association with prominent cranial autonomic features (lacrimation, nasal congestion, rhinorrhea, and conjunctival injection).
Disorders include cluster headache, paroxysmal hemicrania, and SUNCT syndrome.
Cluster headache
Hx: Cluster headaches are characterized by unilateral, severe, boring pain that is usually orbital, supraorbital, and/or temporal in location
The time from onset to peak intensity is usually minutes, with the pain lasting 15 minutes to 3 hours.
Accompanying autonomic symptoms include lacrimation, nasal congestion, rhinorrhea, miosis, ptosis, and conjunctival injection. The attacks occur in clusters that last weeks to months, with remissions lasting months to years.
Often associated with unilateral tearing and nasal congestion or rhinitis. Pain is severe, unilateral, and periorbital. More common in men but relatively uncommon overall.
Tx: 100% Oxygen inhalation delivered via a non-rebreather face mask at a flow rate of 6 to 12 L/min for 10 minutes is often effective in terminating the attack. Subcutaneous sumatriptan and nasal zolmitriptan are also effective in treating a cluster headache. Verapamil can be effective in preventing cluster headaches.
Nifedipine has been shown to be effective, as has prednisone, indomethacin, and lithium. However, the medication should not be given daily, just during the symptomatic period. Ergotamine is generally only helpful in the acute stage—not for prophylaxis.
Prednisone is usually given to abort the cluster; 40 to 60 mg per day is given for weeks and then tapered over a month or two.
In general, oral medications are not helpful including the oral serotonin antagonists.
Subarachnoid hemorrhage
Sudden, explosive onset of severe headache (“worst headache of my life”)[“Thunderclap”] ⚡. Preceded by “sentinel” headache in 10%. (“Berry”) Aneurysm rupture is the most common etiology for subarachnoid bleeds, and smoking is considered the most important modifiable risk factor.
Dx: In a minority of patients with a small amount of blood in the subarachnoid space, computed tomography CT of the head may initially be normal.
The most sensitive diagnostic test is the lumbar puncture (LP). The finding one would expect with a subarachnoid hemorrhage is a yellowing of the fluid from the hemolysis of red blood cells. This is known as xanthochromia. Although xanthochromia may be seen through visual inspection of CSF, where the CSF is held against a bright white light compared to a tube of water, the most sensitive diagnostic method is spectrophotometry. Spectrophotometry detects blood breakdown products as they progress from oxyhemoglobin to bilirubin. The sensitivity is >95% when a lumbar puncture is performed more than 12 hours after the initial hemorrhage.
Tx: Medical treatment is directed toward preventing elevation of arterial or intracranial pressure that might re-rupture the aneurysm or AVM. Typical measures include absolute bed rest with the head of the bed elevated 15 to 20 degrees, mild sedation, and analgesics for headache (antiplatelet drugs should be avoided). Because patients who are hypertensive on admission have increased mortality, reducing blood pressure (to approximately 160-170/100 mm Hg) is prudent, but bed rest and mild sedation are often adequate in this regard. Cx: Pressure not too low because some of the elevated pressure may represent a compensatory mechanism to maintain cerebral perfusion pressure in the face of increased intracranial pressure or cerebral arterial narrowing. Tx: IV nitroprusside 🧨is a good agent to use because it can be titrated with the blood pressure. If the pressure drops too low, the IV can be turned off.
Cx: Rebleeding is the major cause of death within the first 24 hours of presentation, especially within the first 6 hours of untreated SAH. Vasospasm can occur in up to 30% of SAH patients from days 3-10 after presentation and is the major cause of delayed morbidity and death. It is likely caused by arterial narrowing at the base of the brain due to degradation of the blood and its metabolites and can lead to cerebral infarction. CT angiography is preferred for detecting vasospasm, which can best be prevented with initiation of nimodipine.
CSF Leak
Post–lumbar-puncture headache is diagnosed by a history of a dural puncture (eg, spinal tap, spinal anesthesia) and is characteristically a postural headache, with marked increase in pain in the upright position and relief with recumbency. The pain is typically occipital, comes on within 48 to 72 hours after the procedure, and lasts 1 to 2 days. Nausea and vomiting may occur. Headache is caused by persistent leak of CSF from the spinal subarachnoid space, with resultant traction on pain-sensitive structures at the base of the brain. The risk of this complication can be reduced by using a small-gauge needle (22 gauge or smaller) for the puncture. Lying flat afterward, for any length of time, does not lessen the risk. Low-pressure headache syndromes are usually self limited.
Cerebrospinal fluid (CSF) rhinorrhea or otorrhea - Leakage of CSF from the nose or ear. CSF rhinorrhea must be distinguished from other causes of rhinorrhea, such as allergic rhinitis. Glucose concentration does not reliably distinguish CSF from nasal mucus, but beta-2 transferrin is unique to CSF, and its presence documents a CSF source of rhinorrhea.
Trigeminal Neuralgia (tic douloureux)
Hx: Characterized by usually unilateral, severe pain in the face in the distribution of the maxillary (V2) and/or mandibular (V3) branches of the fifth cranial nerve (trigeminal nerve). It typically has a stabbing or electric shock-like nature, which lasts briefly for a few seconds and can recur several times. It can be accompanied by a brief facial spasm.
Multiple sclerosis (MS) is one of the few conditions that may present with trigeminal neuralgia bilaterally. This occurs due to demyelination of the nucleus of the trigeminal nerve or the nerve root, which leads to improper signaling of the nerve and paroxysms of severe pain.
Px: Physical examination is typically normal and with the typical history and in the absence of any physical findings,
Tx: It is appropriate to initiate therapy with carbamazepine, which is considered the treatment of choice. In the presence of any neurologic signs on examination, imaging and other studies should be obtained to rule out secondary causes. Other medications which can be effective include phenytoin, gabapentin and baclofen. Surgical decompression can be considered in refractory cases.
Giant cell (temporal) arteritis
Systemic symptoms
- Fever, fatigue, malaise, weight loss
Localized symptoms
- Headaches: Located in temporal areas
- Jaw claudication: Most specific symptom of GCA
- PMR
- Arm claudication: Associated bruits in subclavian or axillary areas
- Aortic wall thickening or aneurysms
- CNS: TIAs/stroke, vertigo, hearing loss
Visual symptoms
- Amaurosis fugax: Transient vision field defect progressing to monocular blindness
- Anterior ischemic optic neuropathy (AION): Most common ocular manifestation
Anterior ischemic optic neuropathy is the most common ocular manifestation and is detected on funduscopy by the presence of a swollen and pale disc with blurred margins.
Laboratory results
- Normochromic anemia
- Elevated ESR & CRP
- Temporal artery biopsy
Treatment
- PMR only: Low-dose oral glucocorticoids (eg, prednisone 10-20 mg daily)
- GCA: Intermediate- to high-dose oral glucocorticoids (eg, prednisone 40-60 mg daily)
- GCA with vision loss: Pulse high-dose IV glucocorticoids (eg, methylprednisolone 1,000 mg daily) for 3 days followed by intermediate- to high-dose oral glucocorticoids
Giant cell (temporal) arteritis produces inflammatory changes that affect branches of the external carotid, cervical internal carotid, posterior ciliary, extracranial vertebral, and intracranial arteries
Occurs almost exclusively in patients aged >50 y. Associated with tenderness of the scalp and temporal artery, jaw claudication (muscular pain due to inadequate blood flow), and visual changes (red flag).
Px: May show tender, nodular, or pulseless temporal arteries.
Dx: Laboratory findings include an increased erythrocyte sedimentation rate and evidence of vascular stenosis or occlusion on angiography or color duplex ultrasonography.
Benign (Idiopathic) Intracranial Hyprtension
Risk factors
- Obese women of childbearing age
- Medications (eg, retinoids, tetracyclines, growth hormone)
Excessive vitamin A and its metabolites (eg, isotretinoin) are believed to impair cerebrospinal fluid reabsorption, leading to increased intracranial pressure (ICP).
Clinical features
- Headache
- Vision loss; enlarged blind spot
- Pulsatile tinnitus
- Diplopia; palsy of the abducens nerve (CN VI) [lateral rectus palsy]
- Papilledema
Diagnosis
- Neuroimaging
- Lumbar puncture: elevated opening pressure
Treatment
- Weight loss
- Acetazolamide
Brain parenchyma, cerebrospinal fluid, and blood are contained within a rigid skull and increased volume of any of these 3 components may cause ICH. Potential etiologies include trauma, space-occupying lesions, hydrocephalus, impaired central nervous system venous outflow, and idiopathic ICH (pseudotumor cerebri).
Hx: Patients typically present with headache (worse at night), nausea/vomiting, and mental status changes (eg, decreased level of consciousness, cognitive dysfunction). Patients may also have focal neurologic symptoms (eg, vision changes, unsteady gait) and seizure. Symptoms can worsen with maneuvers that further increase intracranial pressure (eg, leaning forward, Valsalva, cough).
Px: Signs may include papilledema and focal neurologic deficits. Cushing reflex (hypertension, bradycardia, respiratory depression) is a worrisome finding suggestive of brainstem compression.
Benign intracranial hypertension (pseudotumor cerebri)
The pathology involves impaired absorption of CSF by the arachnoid villi.
Hx: Patients with IIH typically present with holocranial headache, vision changes (blurry vision and diplopia), and pulsatile tinnitus (“whooshing” sound in the ears). Although IIH is frequently seen in young obese women, it has also been associated with certain medications (eg, isotretinoin, tetracyclines, growth hormone, excessive vitamin A). Can lead to blindness.
Px: Neurologic examination is normal but may reveal sixth cranial nerve palsy. Headache aggravated by coughing, straining, or changing position. Cerebrospinal fluid pressure is elevated.
Dx: Initial evaluation of IIH includes complete ocular examination and neuroimaging to exclude secondary causes of intracranial hypertension (eg, mass, hemorrhage, cerebral vein thrombosis). Magnetic resonance imaging (MRI) possibly with magnetic resonance venography (to rule out cerebral vein thrombosis) is the preferred imaging modality. Empty sella is present in about 70% of patients but is not diagnostic. Lumbar puncture (LP) is then indicated to document elevated opening pressure.
Increased intracranial pressure (ICP) on the optic disc causes swelling (papilledema), leading to blurry vision that does not improve with refraction and an enlarged blind spot. On funduscopic examination, the optic disc appears elevated with blurred margins, and vessels may be engorged or leaky (ie, splinter hemorrhages). In young children, increased ICP is more likely to affect the cranial nerves, particularly the abducens nerve (CN VI) because of its long course through the skull to the lateral rectus muscle. Patients with CN VI palsy may have diplopia and impaired eye abduction on examination.
❗ Papilledema is NOT a contraindication to LP unless the patient has evidence of obstructive or noncommunicating hydrocephalus and/or a space-occupying lesion with/without mass effect or midline shift.
Pseudotumor cerebri causes a communicating hydrocephalus (ie, pressures in the ventricular and subarachnoid spaces are equilibrated with the lumbar cistern) and therefore LP is considered safe. LP is performed with the patient in the lateral decubitus position with legs extended. An opening pressure of >250 mm H2O is considered abnormal and in the appropriate clinical setting is diagnostic of IIH. Cerebrospinal fluid studies are normal in IIH patients.
Tx:
The treatment includes weight reduction and acetazolamide (if weight reduction fails). When medical measures fail or visual field defects are progressive, shunting or optic nerve sheath fenestration is done to prevent blindness, which is the most significant complication of this otherwise benign disorder.
Acetazolamide is the first-line medical treatment for IIH. It inhibits choroid plexus carbonic anhydrase, thereby decreasing CSF production and IH.
Furosemide can be added for patients with continued symptoms on acetazolamide.
Toxoplasmosis
Fever, headache, focal neurologic deficits; multiple ring-enhancing lesions on CNS imaging; positive toxoplasma serology
All patients newly diagnosed with HIV should be tested for latent infection with serology for T gondii IgG antibody. If serology is positive and CD4 count is <100/mm3, primary prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX) reduces the risk of toxoplasmosis dramatically (to 0%-2%).
Patients on antiretroviral treatment can discontinue TMP-SMX when CD4 count is >200/mm3 for 3 months (and there is adequate viral suppression). TMP-SMX is also used for primary prophylaxis against Pneumocystis pneumonia.
🖌 STROKE
- Stroke
- TIA
- Seizure
- Hypoglycemiacor
- Complicated migraine
- Mass lesion (tumor, abscess, subdural hematoma)
- Encephalitis
- Functional
Clinical characteristics of major stroke subtypes
Ischemic (thrombotic)
- Atherosclerotic risk factors (eg, uncontrolled hypertension, diabetes), ± history of transient ischemic attack
- Local obstruction of an artery (eg, carotid, cerebral, vertebral)
- Symptoms may alternate with periods of improvement (stuttering progression)
Ischemic (embolic)
- History of cardiac disease (eg, atrial fibrillation, endocarditis) or carotid atherosclerosis
- Onset of symptoms is abrupt & usually maximal at the start
- Multiple infarcts in different vascular territories
Intracerebral hemorrhage
- History of uncontrolled hypertension, coagulopathy, illicit drug use (eg, amphetamines, cocaine)
- Symptoms progress over minutes to hours
- Focal neurologic symptoms appear early, followed by features of increased intracranial pressure (eg, vomiting, headache, bradycardia, reduced alertness)
Spontaneous subarachnoid hemorrhage
- Bleeding from arterial saccular (“berry”) aneurysm or arteriovenous malformation
- Severe headache at onset
- Meningeal irritation (eg, neck stiffness)
- Focal deficits uncommon
TIA
An episode of focal cerebral ischemia that resolves fully and rapidly, usually within 1 hour, without evidence of cerebral infarction.
Hx: Same clinical features as for stroke.
Dx: ABCD2 score assigns one point for an Age of 60 years or greater, one point for a Blood pressure of 140/90 mm Hg or greater, two points for the Clinical symptom of hemiparesis, two points for Duration of 60 minutes or greater, and one point for the presence of Diabetes mellitus). The American Heart Association guidelines recommend hospital admission for all patients with probable TIAs whose ABCD2 scores are 3 or greater.
Tx: The goal of treatment is to prevent subsequent stroke, which occurs in up to 10% of patients in 2 days and up to 20% of patients in 90 days.
Discontinuing tobacco use, initiating 🧯aspirin, starting a statin for hyperlipidemia, and reducing blood pressure.
Ischemic Stroke / Aphasia
The underlying pathologic process in stroke can be either ischemia or hemorrhage, usually arising from an arterial lesion. Ischemia (90%) and hemorrhage (10%) of strokes.
Among ischemic strokes:
50% are attributed to cardioembolism
25% to large artery occlusion: gaze palsy, aphasia, neglect
10% to small artery occlusion
Remainder of unknown origin or cryptogenic [TOAST trial].
Hx: Thrombotic stroke: Often presents with stepwise progression of neurologic deficits and may be preceded by one or more TIAs with identical symptoms.
Lacunar infarcts are also characteristically thrombotic.
Cardioembolic stroke is suggested by maximal deficit within 5 minutes of onset, impaired consciousness at onset.
Px: Blood Pressure for elevation, blood pressure comparison, ophthalmoscopic examination for ebolization of the anterior circulation, Neck examination for the absence of carotid pulses or bruit, cardiac examination for murmurs, skin exam for ecchymoses or petichiae.
Aphasia, unilateral neglect or constructional apraxia suggest a cortical lesion in the anterior circulation and exclude vertebrobasilar or lacunar stroke.
Coma implies brainstem or bihemispheric involvement.
Visual Field (Hemianopia) can occur with occlusion of either the middle or posterior cerebral artery, which supply the optic radiation and visual cortex, respectively.
Isolated hemianopia suggests posterior cerebral artery stroke, because middle cerebral artery stroke should produce additional (motor and somatosensory) deficits.
Ocular palsy, nystagmus, or internuclear ophthalmoplegia assigns the underlying lesion to the brainstem and thus the posterior cerebral circulation.
Hemiparesis can be due to lesions in cerebral cortical regions supplied by the anterior circulation, descending motor pathways in the brainstem supplied by the vertebrobasilar system, or lacunes at subcortical or brainstem sites.
Crossed hemiparesis, which involves the face on one side and the rest of the body on the other, assigns the lesion to the brainstem between the facial (VII) nerve nucleus in the pons and the decussation of the pyramids in the medulla.
Cortical sensory deficits such as astereognosis or agraphesthesia, with preserved primary sensory modalities, imply a cortical deficit within the middle cerebral artery territory.
Hemiataxia usually points to a lesion in the ipsilateral brainstem or cerebellum but can also be produced by lacunar stroke in the internal capsule.
Dx: Serum glucose/HbA1c, creatinine and lipid profile (risk factors), CBC (thrombocytosis, polycythemia, hemoglobin electrophoresis (sickle cell), infection [blood cultures]), coags, ESR/ANA (vasculitis), VDRL (syphilitic arteritis), Troponin (ischemia). ECG/telemetry (MI, afib), LP (subarrachnoid hemorrhage), PT/INR (antigoagulation anticipation), Lupus anticoagulant, anticardiolipin antibody, factor V Leiden, protein C, protein S, antithrombin III (hypercoagulable states), LDL (hypercholesterolemia), TTE (hypokinesis/shunt).
Noncontrast CT is usually preferred for initial diagnosis because it is widely available and rapid and can readily make the critical distinction between ischemia (-) and hemorrhage (+).
Duplex (doppler) ultrasonography of the carotid arteries should be performed within the first 2 days to assess for stenosis warranting consideration of carotid endarterectomy (and is noninvasive),
DWI is superior to CT for detecting stroke during the first 12 hours after onset and may help predict final infarct volume in anterior circulation stroke, although diffusion defects are sometimes seen with TIAs, and small strokes or brainstem strokes may escape detection. The difference between DWI and PWI abnormalities (diffusion-perfusion mismatch) may represent tissue that is at risk of infarction but potentially salvageable by thrombolysis, which equates roughly to the ischemic penumbra.
Magnetic resonance angiography (MRA) is a noninvasive substitute for digital subtraction angiography and can detect extracranial carotid disease with high sensitivity and specificity.
CT angiography (CTA) is an alternative, but involves radiation exposure and may be obscured by artifact from calcium in atherosclerotic plaques.
Tx:
Blood pressure should usually not be lowered acutely, except for patients with acute ischemic stroke in whom it is high enough (>185 mm Hg systolic or >110 mm Hg diastolic pressure) to make an otherwise suitable candidate ineligible for thrombolytic therapy.
🎨 Interventional
Thrombolysis (3-4.5 h): Alteplase (recombinant tissue-type plasminogen activator) increases the chance of recovery from ischemic stroke when administered intravenously within 4.5 hours of symptom onset OR within 3 hours of when the patient was last seen awake and without symptoms. Brain imaging must be negative for hemorrhage.
Strict exclusion criteria
- Hemorrhage or multilobar infarct involving >33% of cerebral hemisphere on CT scan
- Stroke/head trauma in the past 3 months
- History of intracranial hemorrhage, neoplasm, or vascular malformation
- Recent intracranial/spinal surgery
- Active bleeding or arterial puncture in the past 7 days at noncompressible site
- Blood pressure >185/110 mm Hg
- Platelets <100,000/mm3 or glucose <50 mg/dL
- Anticoagulant use with INR >1.7, PT >15 sec, or ↑ active PTT
Relative exclusion criteria
- Minor or rapidly improving neurodeficits
- Major surgery/trauma in past 14 days
- Myocardial infarction in the past 3 months
- GU or GI bleeding in the past 21 days
- Seizure at stroke onset
- Pregnancy
❗ Contraindications are designed to avert unnecessary or ineffectual treatment include the presence of only minor or resolving neurologic deficits; onset of symptoms more than 6 hours prior to initiating treatment; and hypoglycemia (blood glucose <50 mg/dL), which can mimic stroke.
Related to bleeding complications: recent head trauma > 3 months, major surgery in the last 2 weeks, or hx of hemorrhage, stroke within 1 month; blood pressure greater than 185 mm Hg systolic or greater than 110 mm Hg diastolic pressure; and impaired coagulation (INR >1.7, elevated aPTT, or platelet count 14 days postpartum.
Within the first 24 hour after administration of rtPA, anticoagulants and antiplatelet agents should not be given, blood pressure should be carefully monitored, and arterial puncture and placement of central venous lines, bladder catheters, and nasogastric tubes should be avoided.
Rx: Intravenous alteplase (tissue plasminogen activator) has been shown to improve neurologic outcomes in patients with ischemic stroke when given within 4.5 hours of symptom onset.
0.9mg/kg over 60 mins (do not exceed 90 mg total dose)
T M O N A B A S H
Stroke with no prior antiplatelet therapy
- 🧯 Aspirin
Stroke on aspirin therapy
- Aspirin ➕ dipyridamole OR clopidogrel
Stroke with evidence of atrial fibrillation
- Long-term anticoagulation (eg, warfarin, dabigatran, rivaroxaban)
Patient with intracranial large-artery atherosclerosis
- Aspirin + clopidogrel for 90 days, then aspirin
Stroke with large anterior circulation artery occlusion within 24 hours of symptom onset
- Mechanical thrombectomy (regardless if patient received alteplase), then aspirin
Mechanical thrombectomy with stent or coil retrievers. Okay up to 8-12 hours, depending on brain tissue viability (collateralization), NIHSS score and vessel accessibility.
🔪 Surgical:
Carotid endarterectomy (surgical removal of thrombus from a stenotic common or internal carotid artery in the neck) is indicated for patients with anterior circulation TIAs and high-grade (70%-99%) extracranial internal carotid artery stenosis
Carotid artery stenting is as effective as endarterectomy for treating extracranial carotid stenosis, assuming a similar perioperative morbidity and mortality rate. Stenting is associated with an increased risk of periprocedural stroke, but a decreased risk of periprocedural myocardial infarction.
NIH Stroke Scale (NIHSS) Predicts lieklihood of recovery after stroke; >16 high = high risk of death or severe disability at 3 months; <6 = high likelihood of good recovery at 3 months.
✅ ICA Branches and syndromes
Ophthalamic:
Central retinal artery - sudden ipsilateral painless vision loss.
Ciliary arteries - inability to focus visually
Anterior Choroidal
- Contralateral hemiplegia (internal capsule)
- Homonymous hemianopsia (optic tracs and LGN)
- Movement problems (striatum)
Posterior Communicating (thalamic problems, Aneurysm/CNIII palsy, hemiparesis)
Anterior cerebral artery: stroke produce contralateral paralysis and sensory loss exclusively or primarily affecting the 🦵leg. There may also be abulia (lack of will or initiative), disconnection syndromes such as the alien hand (involuntary performance of complex motor activity), transcortical expressive aphasia and urinary incontinence.
🔴 Middle Cerebral Artery:
Superior division stroke results in contralateral hemiparesis that affects the face, hand, and arm. If the dominant hemisphere is involved, there is:
Broca (expressive) aphasia (L post. inf. frontal lobe), which is characterized by impaired language expression (📝writing, repeating, fluency), intact comprehension and reading; sensory loss (post-central gyrus) [dominant hemisphere]
Wernicke’s difficulty comprehending and following commands but are able to speak fluently. However, their speech tends to be rambling without concrete meaning.
Aprosodia: MCA; non-dominant hemisphere: Monotonus, flat speech for anterior; inability to comprehend tone of speech for posterior.
Hemineglect: MCA, parietal cortex: on non-dominant hemisphere
Nondominant parietal lobe lesions typically cause anosognosia (denial of one’s disabilities) and contralateral apraxia (inability to carry out learned purposeful movements)
Dominant temporal lobe lesions can affect comprehension (receptive aphasia), ability to speak nouns (anomic aphasia), repetition (conductive aphasia) due to arcuate fasciculus involvement, and contralateral superior homonymous quadrantanopsia due to inferior optic radiations (Meyer’s loop) involvement.
Nondominant temporal lobe lesions can impair ability to comprehend emotional gestures (sensory aprosodia).
Inferior division stroke results in contralateral homonymous hemianopia (LGN) that may be denser inferiorly, quadrantanopsia (Myer’s loop), impaired cortical sensory functions (eg, graphesthesia and stereognosis) on the contralateral side of the body, and disorders of spatial thought (eg, anosognosia [unawareness of deficit], neglect of the contralateral limbs and contralateral side of external space, dressing apraxia, and constructional apraxia). If the dominant hemisphere is involved:
Wernicke’s [L. post. sup. temp. lobe](receptive) aphasia (sup. temp.) occurs and is manifested by impaired comprehension, reading, and repeating, and fluent and can write, but often nonsensical speech.
Conduction aphasia occurs in the left arcuate fasciculus region, and gives fluent spontaneous speech, good auditory comprehension, and poor repetition and naming.
Global aphasia occurs from damage to the left perisylvian region, and as the name suggests, gives a nonfluent aphasia with poor auditory comprehension, repetition, and naming.
Anomic aphasia occurs in the left angular gyrus, and affected
individuals have fluent spontaneous speech, good auditory comprehension and repetition, and poor naming.
Occlusion of the stem of the middle cerebral artery occurs proximal to the origin of the lenticulostriate branches, resulting in a clinical syndrome similar to that seen after occlusion at the trifurcation (both above). In addition, however, involvement of the internal capsule causes paralysis of the contralateral leg, so hemiplegia and sensory loss affect face, hand, arm, and leg.
M1 segment - segments of the frotnal, sup. temp., insular, parietal, corona radiata, putamen, internal capsule, globus pallidus.
Gerstmann syndrome: Constellation of acalculia, finger agnosia, right-left confusion and agraphia, occurs with damage to the dominant inferior parietal lobe. MCA on dominant inf. parietal lobe.
Astereognosia: MCA dominant somatosensory association cortex (inability to indentify objects by feel)
Agraphesthesia: MCA dominant somatosensory association cortex (inability to identify letters written on hand).
M2 segment - Language, and other areas surrounding sylvian fissue.
M3 segment - cortical areas (pre and post central gyrus).
🔴Posterior cerebral artery
- Occlusion produces homonymous hemianopia affecting the contralateral visual field, except that macular vision may be spared.
🔴Superior cerebellar artery occlusion
Causes lateral rostral pontine infarction and resembles anterior inferior cerebellar artery lesions, but impaired optokinetic nystagmus or skew deviation of the eyes may occur, auditory function is unaffected, and the contralateral sensory disturbance may involve touch, vibration, and position sense as well as pain and temperature sense.
🔴 Basilar thrombosis usually affects the proximal basilar artery which supplies the pons.
Involvement of the dorsal pons (tegmentum) produces unilateral or bilateral abducens (VI) nerve palsy; horizontal eye movements are impaired, but vertical nystagmus and ocular bobbing may be present.
If the ventral pons (basis pontis) is infarcted and the tegmentum is spared, patients remain conscious but quadriplegic (locked-in syndrome). Locked-in patients may be able to open or move their eyes vertically on command.
Medial Pontine syndrome causes contralateral spastic hemiparesis (corticospinal tract), contralateral loss of vibration and position (ML).
Lateral Pontine syndrome produces symptoms similar to wallenberg except also have facial droop (VII) and hearing loss (VIII). Not vertigo. Anterior inferior cerebellar artery occlusion leads to infarction of the lateral portion of the caudal pons and produces ipsilateral facial weakness, gaze palsy, deafness, and tinnitus.
🔴Posterior inferior cerebellar artery occlusion
Lateral medullary (Wallenberg) syndrome
Loss of pain/temp over the ipsilateral face (spinal trigeminal), contralateral body (spinothalamic), ipsilateral bulbar (gag, dysphagia, hoarseness), muscle weakness (nucleus ambiguous), vertigo (vestibular nucleus), ipsilateral limb ataxia (inferior cerebellar peduncle), and horner syndrome (descending sympathetic fibers).
+Nystagmus, nausea, vomiting, dysarthria, and hiccup.
🔴Anterior Spinal Artery occlusion
Produces medial medullary syndrome. Patients present with hemiparesis (corticospinal tract) contralateral hemisensory loss (vibration/position) (DCML), and ipsilateral tongue pralysis (hypoglossal nucleus).
Lacunar infarcts
Lacunar strokes are small (<15 mm in diameter) subcortical infarcts resulting from occlusion of deep penetrating branches of the major cerebral arteries (eg, anterior cerebral, middle cerebral, basilar). Affected areas typically include the basal ganglia, subcortical white matter (internal capsule, corona radiata), and pons. Lacunar infarcts are most commonly associated with chronic ♨ hypertension, which leads to arteriolar sclerosis and vessel occlusion (hypertensive vasculopathy). Other risk factors include diabetes, smoking, advanced age, and increased LDL cholesterol.
Lacunar infarcts typically produce neurologic deficits over minutes to hours and symptoms may follow a stuttering course.
The absence of cortical signs (eg, aphasia, agnosia, neglect, apraxia, hemianopia), seizures, and mental status changes (eg, stupor, coma) also supports a deep/subcortical localization.
🔼 Lacunar Infarction Stem from the lenitculostriate branches of the middle cerebral artery (MCA) supply the internal capsule, ACA (recurrent artery of hubner), or the basilar artery (paramedian branches) affecting the putamen, globus pallidus, less commonly the thalamus (supplied by penetrating arteries from the PCA), caudate nucleus, pons, posterior limb of the internal capsule.
There are five classic and distinctive lacunar syndromes:
Pure motor hemiparesis — Characterized by weakness involving the face, arm, and leg on one side of the body in the absence of “cortical” signs (aphasia, agnosia, neglect, apraxia, or hemianopsia) or sensory deficit. Lacunes that produce this syndrome are usually located in the contralateral internal capsule or pons. Pure motor hemiparesis also may be caused by internal carotid or middle cerebral artery occlusion, subdural hematoma, or intracerebral mass lesions.
Pure sensory stroke — Defined as numbness of the face, arm, and leg on one side of the body in the absence of motor deficit or “cortical” signs. Results from lacunar infarction in the contralateral thalamus. It may be mimicked by occlusion of the posterior cerebral artery or by a small hemorrhage in the thalamus or midbrain. Several weeks to months following the stroke, sensory deficits can improve; however, some patients develop thalamic pain syndrome (Dejerine-Roussy syndrome). This condition is characterized by severe paroxysmal burning pain over the affected area and is classically exacerbated by light touch (allodynia).
Ataxic hemiparesis — [sometimes called ipsilateral ataxia and crural (leg) paresis]. Patients characteristically develop ipsilateral weakness and limb ataxia that is out of proportion to the motor deficit and usually affects the leg predominantly. Symptoms result from a lesion in the contralateral pons, internal capsule, or subcortical white matter.
Sensorimotor stroke — Sensorimotor stroke is characterized by weakness and numbness of the face, arm, and leg on one side of the body in the absence of the aforementioned “cortical” signs. Arise from infarcts involving the posterolateral thalamus AND posterior limb of the internal capsule.
Dysarthria-clumsy hand syndrome — Facial weakness, dysarthria, dysphagia, and slight weakness and clumsiness of one hand are characteristic. There are no sensory deficits or “cortical” signs. Least common. Lacunes causing this syndrome are located in the contralateral pons or internal capsule.
Internal Capsule: Posterior Limb: Contralateral hemiparesis and hemianesthesia (due to disruption of the corticospinal and somatosensory fibers)
Internal Capsule: Anterior Limb: conjugate gaze deviation toward the side of the lesion (due to damage of frontal eye field efferents).
Hemorrhagic Stroke
Intracerebral hemorrhage tends to cause more severe headache and depression of consciousness as well as neurologic deficits that do not correspond to the distribution of any single blood vessel.
Hemorrhage usually occurs during routine activity and most often involves the occipital and parietal lobes. Parietal hemorrhages can cause contralateral hemisensory loss (due to primary somatosensory cortex injury) and contralateral hemineglect if the parietal association cortex (particularly in the nondominant hemisphere) is affected.
Arteriovenous malformation rupture is the most common cause of intracranial hemorrhage in children 👶🏼
Amyloid angiopathy is the most common cause of spontaneous lobar hemorrhage, particularly in adults age >60. It occurs as a consequence of β-amyloid deposition in the walls of small- to medium-size cerebral arteries, resulting in vessel wall weakening and predisposition to rupture. The disease is NOT associated with systemic amyloidoses; rather, the amyloidogenic proteins are usually the same as those seen in Alzheimer dementia.
Hypertensive hemorrhages generally involve the same small, penetrating arteries that are responsible for lacunar stroke. The most frequently affected locations include the basal ganglia (putamen), cerebellar nuclei, thalamus, and pons.
Putaminal hemorrhage almost always involves the adjacent internal capsule. This leads to contralateral hemiparesis and hemianesthesia (due to disruption of the corticospinal and somatosensory fibers in the posterior limb) and conjugate gaze deviation toward the side of the lesion (due to damage of frontal eye field efferents in the anterior limb).
Cerebellar hemorrhage presents with occipital headache, nausea/vomiting, dizziness, and cerebellar signs (eg, ataxia, dysmetria).
Thalamus: upgaze palsy;
Pons: pinpoint pupils
Tx: Treat fever; insulin to treat hyperglycemia; compression devices; avoid hypotonic fluids, anticoagulation reversal (vitamin K (Warfarin), protamine sulfate (heparin), reversal agents);
⛑ Warfarin-associated intracerebral hemorrhage: Initial therapy should include IV vitamin K, which has a sustained response but takes approximately 12-24 hours to be effective (promotes clotting factor synthesis in the liver).
Prothrombin complex concentrate (PCC) should also be provided as it contains vitamin K-dependent clotting factors (eg, II, VII, IX, X) that offer rapid (minutes) and short-term (hours) reversal of warfarin.
Fresh frozen plasma can be considered if PCC is not available; however, it takes longer to prepare/administer and requires more volume infusion compared to PCC.
Traget BP <140 within 1 hour (INTERACT study);
For increased ICP: head of the bed to 30 degrees; hyperventilation, mannitol/hypertonic saline, sedation, monitor cerebral perfusion pressure; IVC placement, seizure prophylaxis, surgery (craniotomy, hematoma evacuation, ventruculostomy).
Complicated migraine
Hx: Suspect in younger patients, more often women with history of severe headache. Similar onset and focal findings as in stroke. Usually severe headache preceding or following attack.
Px: Sensory and visual disturbances often prominent; sensory symptoms often spread over affected area.
Dx: MRI usually normal.
Hemiplegic Migrane
Hx: Ocurrence of episodic, reversible motor weakness as a manifestation of migraine aura in conjunction with at least one other kind of aura (visual, sensory, aphasic, or brainstem).
At least two of the following four characteristics:
- At least one aura symptom spreads gradually over ≥5 minutes, and/or two or more symptoms occur in succession
- Each individual non-motor aura symptom lasts 5 to 60 minutes, and motor symptoms last <72 hours
- At least one aura symptom is unilateral
- The aura is accompanied, or followed within 60 minutes, by headache
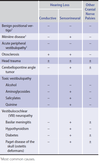
⛑ Bacterial meningitis
Hx: Fever, severe headache, stiff neck, photophobia, drowsiness or confusion, nausea, vomiting.
S. pneumoniae is the most common cause and may occur in patients with other foci of infection (eg, pneumonia, otitis media, mastoiditis, sinusitis, or endocarditis) or following head trauma with leakage of cerebrospinal fluid (CSF).
N. meningitidis is the second most common cause of bacterial meningitis in the United States, occurring primarily in children and young adults.
L. monocytogenes meningitis develops most frequently in neonates, older adults (>50 years of age), and those who are immunocompromised (diabetes mellitus, liver or kidney disease, collagen vascular disorders, disorders of iron overload, HIV infection, transplant recipients, and patients taking anti-tumor necrosis factor α agents such as infliximab and etanercept)
Dx: CSF findings that predict bacterial etiology with ≥99% certainty include:
Protein concentration >220 mg/dL (2200 mg/L)
Glucose concentration <34 mg/dL (1.9 mmol/L)
CSF-blood glucose ratio <0.23
Leukocyte count >2000/µL (2 x 109/L)
Neutrophil count >1180/µL (1.18 x 109/L)
A computed tomography (CT) scan of the head should be done before lumbar puncture in mass lesion suspected patients, as well as patients who are immunocompromised, have a history of CNS disease, present with new-onset seizures, or have a decreased level of consciousness, focal neurologic deficits, or papilledema.
Tx:VancomycinAND anIV third-generation cephalosporin(eitherceftriaxoneorcefotaxime) (ANDampicillin in adults age >50) are empiric treatments for bacterial meningitis.




and interfering with daily functioning and independent living.

