Haem: Myelodysplastic syndromes and Aplastic anaemia Flashcards
Define myelodysplastic syndrome.
Biologically heterogenous group of acquired haematological stem cell disorders
What are the key characteristics of myelodysplastic syndromes?
Development of a clone of marrow stem cell with abnormal maturation resulting in
- Functionally defective blood cells
- Numerial reduction in cell counts
This leads to
1. Cytopaenia
2. Functional abnormalities of erythroid, myeloid, and megakaryocyte maturation
3. Increased risk of transformation to leukaemia
Which types of patients tend to develop myelodysplastic syndromes?
Elderly
How do myelodysplastic syndromes typically present?
Symptoms/signs of bone marrow failure developing over weeks/months
List and describe some blood and bone marrow features of myelodysplastic syndromes - seen on blood film
- Pelger-Huet anomaly - bilobed neutrophils
- Dysgranulopoeisis of neutrophils - failure of granulation
- Dyserythropoiesis of red blood cells -lack of separation between red cell precursors (cytoplasmic bridge), presence of abnormal ring of cytoplasm around the nucleus of percursor red cells
- Dysplastic megakaryocytes - micro-megakaryocytes
- Increased proportion of blast cells in the bone marrow (normally < 5%)
What does this image show?

Pelger-Huet anomaly
Nucleus appears bilobed or dumb-bell shaped
Normal neutrophils shown below

What does this image show?

Refractory anaemia with dysgranulopoiesis - failure of neutrophil granulation

What does this image show?

Refractory anaemia-dyserythropoiesis
- Lack of separation between red cell precursors
- Presence of abnormal blebby ring of cytoplasm around the nucleus of precursor red cells

What does this image show?
Name the stain used

Ringed sideroblasts
- Accumulation of iron around the nuclei of red blood cell precursors
- Seen in refractory anaemia with ringed sideroblasts (RARS)
Seen on Prussian blue stain
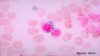
What is the presence of myeloblasts with Auer rods suggestive of?
Acute myeloid leukaemia.

List the criteria of the WHO classification of MDS

What are the five prognostic variables that are used to calculate prognostic risk using the Revised International Prognostic Scoring System (IPSS-R) for Myelodysplastic Syndromes?
The high risk is considered a score > 6, low risk ≤ 1.5
The higher the risk, the lower the survival and time to progress to AML

How does myelodysplasia tend to evolve from the time of diagnosis?
- Blood count deterioration - leads to worsening bone marrow failure)
- Development of acute myeloid leukaemia - extremely poor prognosis
What are the usual causes of death in patients with myelodysplasia?
- 1/3 infection
- 1/3 bleeding
- 1/3 leukaemia
What are the two treatments that can prolong life in myelodysplastic syndromes?
- Allogeneic stem cell transplantation
- Intensive chemotherapy
NOTE: as most MDS patients are elderly, they often cannot tolerate treatment
List some other treatments that may be used in myelodysplastic syndromes.
Supportive Care
- Blood products support
- Antimicrobials
- Growth factors e.g. EPO, G-CSF, TPO-agonist (eltrombopag)
Biological modifiers - treating underlying disease
- Immunosuppression
- Azacytidine or Decitabine (hypomethylating agent)
- Lenalidomide (used in del(5q) minus syndrome)
Oral chemotherapy (e.g. hydroxyurea) - if WCC is high (less common)
Low-dose chemotherapy (SC low-dose cytarabine) - not used now
What is bone marrow failure a result of?
Results from damage or suppression of stem or progenitor cells
- Damage to pluripotent stem cell - impairs production of ALL peripheral blood cells
- Damage to committed progenitor cells - results in bi- or unicytopenias
List some primary and secondary causes of bone marrow failure

List some causes of primary aplastic anaemia
Acquired: idiopathic aplastic anaemia (MOST COMMON - 70-80%)
Congential (RARE):
- Fanconi anaemia (multipotent stem cell)
- Dyskeratosis congentia
List some secondary causes of aplastic anaemia
- Radiation
- Drugs - cytotoxic drugs (predicable), chloramphenicol, NSAIDS (both idosyncratic)
- Autoimmune - SLE
- Infection - parvovirus B19, hepatitis, HIV
List some drugs that can cause bone marrow failure.
- Cytotoxic drugs (predicatble, dose-dependent)
- Phenylbutazone, Gold salts (idiosyncratic, rare)
- Antibiotics - chloramphenicol, sulphonamides
- Diuretics - thiazide
- Antithyroid drugs - carbimazole
What is aplastic anaemia
Hypocellular anaemia
(cytopenia without tumour etc in bone marrow)
Which age groups are affected by aplastic anaemia?
All age groups can be affected - mainly 15-24 and 60+
NOTE: this is much more rare than MDS
What is the most common cause of aplastic anaemia?
Idiopathic (70-80%)
List some inherited causes of aplastic anaemia.
RARE
- Fanconi anaemia
- Schwachman-Diamond syndrome
- Dyskeratosis Congenita

Outline the possible pathophysiology of idiopathic aplastic anaemia.
Characterised by failure of the bone marrow to produce blood cells
- Either due to an inherent issue with the stem cells or due to autoimmune attack on stem cells - spontaneous of molecular mimicry perhaps triggered by viral infection
What are the symptoms of aplastic anaemia?
- Anaemic symptoms - fatigue, SoB
- Leukopaenic symtpoms - infections
- Thrombocytopenic symptoms - easy bruising, bleeding
How is aplastic anaemia diagnosed and what would you see?
- FBC - cytopaenia
- Bone marrow biopsy - hypocellularity

How can aplastic anaemia be classified?
Severe (SAA) or non-severe (NSAA)
List some differential diagnosis for pancytopaenia and hypocellular marrow.
- Hypoplastic MDS/AML
- Hypocellular ALL
- Hairy cell leukaemia
- Atypical mycobacterial infection
- Anorexia nervosa (fatty replacement of bone marrow)
- ITP (although Hb and RBC will be normal)
What is the Camitta criteria for severe aplastic anaemia?
1) 2 out of 3 peripheral blood features:
- Reticulocytes < 1% (<20 x 10^9/L)
- Neutrophils < 0.5 x 109/L
- Platelets < 20 x 109/L
2) Bone marrow cellularity < 25%
Outline the management approaches used for bone marrow failure.
- Seek and remove cause
- Supportive (blood products, antibiotics, iron chelation)
- Immunosuppressive therapy (anti-thymocyte globulin, steroids, ciclosporin A)
- Drugs that promote bone marrow recovery (oxymetholone, eltrombopag)
- Stem cell transplantation
- Other treatments for refractory cases - alemtuzumab (T cell depletion), high-dose cyclophosphamide
Such a rare disease, sent to specialist centre with individualised therpy
How might the age of the patient influence decisions regarding their management?
- Immunosuppressive and androgens therapies tend to be used in older patients
- Stem Cell Transplant tends to be used in younger patients (80% cure rate)
List some late complications that occur after immunosuppressive therapy for aplastic anaemia.
- Relapse (35% in 15 years)
- Clonal haematological disorders - 20% risk in 10 years (myelodysplasia, leukaemia, paroxysmal nocturnal haemoglobinuria)
- Solid tumours (3% risk)
What is the most common cause of inherited aplastic anaemia?
Fanconi anaemia
What is the inheritance pattern of Fanconi anaemia?
Autosomal Recessive or X-linked Recessive
What do the gene mutations implicated in Fanconi anaemia tend to result in?
- Abnormalities in DNA repair
- Chromosomal fragility (breakage in the presence of in vitro mitomycin and diepoxybutane)
List some somatic abnormalities that are seen in Fanconi anaemia.
- Short stature
- Hypopigmented spots/café-au-lait spots
- Abnormality of thumbs
- Microcephaly or hydrocephaly
- Hypogonadism
- Developmental delay
(somatic abnormalities - those which you can see)

NOTE: these are only present in 70% of patients
List some complications of Fanconi anaemia.
- Aplastic anaemia (90%) - median age 9
- Myelodysplasia
- Leukaemia
- Cancer (epithelial)
- Liver disease
What are the 3 characteristic features of dyskeratosis congenita?
- Bone marrow failure
- Cancer predisposition
- Somatic abnormalities (80%)
What are the triad of somatic features in dyskeratosis congenita?
- Abnormal skin pigmentation
- Nail dystrophy
- Leukoplakia

What are telomeres and what are their main functions?
Telomeres are repetitive DNA sequences found at the end of chromosomes
- Prevent chromosomal fusion or rearrangements during chromosomal replication
- Protect genes at the end of chromosomes from degradation
Maintenance of telomere length is required for the indefinite proliferation of human cells.
Telomere length is reduced in bone marrow failure diseases (and they are especially short in dyskeratosis congenita)
Which genes are involved in dyskeratosis congenita and what are the inheritance patterns?
- X-linked recessive (MOST COMMON) - DKC1 gene (defective telomere functioning)
- Autosomal dominant - TERC gene (encodes RNA components of telomerase)
- Autosomal recessive - no mutation identified
NOTE: abnormal telomeric structure and function is heavily implicated in dyskeratosis congenita
How is dyskeratosis congenita akin to idiopathic aplastic anaemia?
Short telomeres are seen in both

